

Solvent accessible surface area-assessed molecular basis of osmolyte-induced protein stability
- PMID: 39131493
- PMCID: PMC11310836
- DOI: 10.1039/d4ra02576h
Solvent accessible surface area-assessed molecular basis of osmolyte-induced protein stability
Abstract
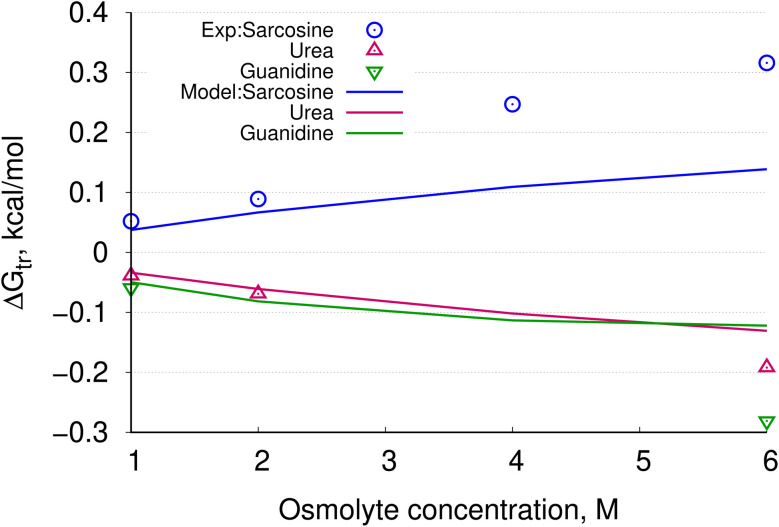
In solvent-modulated protein folding, under certain physiological conditions, an equilibrium exists between the unfolded and folded states of the protein without any need to break or make a covalent bond. In this process, interactions between various protein groups (peptides) and solvent molecules are known to play a major role in determining the directionality of the chemical reaction. However, an understanding of the mechanism of action of the co(solvent) by a generic theoretical underpinning is lacking. In this study, a generic solvation model is developed based on statistical mechanics and the thermodynamic transfer free energy model by considering the microenvironment polarity of the interacting co(solvent)-protein system. According to this model, polarity and the fractional solvent-accessible surface areas contribute to the interaction energies. The present model includes various orientations of participating interactant solvent surfaces of suitable areas. As model systems, besides the backbone we consider naturally occurring amino acid residues solvated in ten different osmolytes, small organic compounds known to modulate protein stability. The present model is able to predict the correct trend of the osmolyte-peptide interactions ranging from stabilizing to destabilizing not only for the backbone but also for side chains. Our model predicts Asn, Gln, Asp, Glu, Arg and Pro to be highly stable in most of the protecting osmolytes while Ala, Val, Ile, Leu, Thr, Met, Lys, Phe, Trp and Tyr are predicted to be moderately stable, and Ser, Cys and Histidine are predicted to be least stable. However, in denaturing solvents, both backbone and side chain models show similar stabilities in urea and guanidine. One of the important aspects of this model is that it is essentially parameter-free and consistent with the electrostatics of the interaction partners that make this model suitable for estimating any solute-solvent interaction energies.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures









Similar articles
-
The amino acid sequence of human chorionic gonadotropin. The alpha subunit and beta subunit.J Biol Chem. 1975 Jul 10;250(13):5247-58. J Biol Chem. 1975. PMID: 1150658
-
Primary structure of murine major histocompatibility complex alloantigens: amino acid sequence of the amino-terminal one hundred and seventy-three residues of the H-2Kb glycoprotein.Biochemistry. 1980 Jan 22;19(2):306-15. doi: 10.1021/bi00543a009. Biochemistry. 1980. PMID: 6986168
-
Primary and tertiary structure of the principal human adenylate kinase.Eur J Biochem. 1976 Sep;68(1):281-90. doi: 10.1111/j.1432-1033.1976.tb10787.x. Eur J Biochem. 1976. PMID: 183954
-
The primary structure of human liver manganese superoxide dismutase.J Biol Chem. 1984 Oct 25;259(20):12595-601. J Biol Chem. 1984. PMID: 6386798
-
The primary structure of iron-superoxide dismutase from Photobacterium leiognathi.J Biol Chem. 1987 Jan 25;262(3):1001-9. J Biol Chem. 1987. PMID: 3542995
Cited by
-
Helix Folding in One Dimension: Effects of Proline Co-Solvent on Free Energy Landscape of Hydrogen Bond Dynamics in Alanine Peptides.Life (Basel). 2025 May 19;15(5):809. doi: 10.3390/life15050809. Life (Basel). 2025. PMID: 40430235 Free PMC article.
References
-
- Yancey P. H. Clark M. E. Hand S. C. Bowlus R. D. Somero G. N. Living with water stress: evolution of osmolyte systems. Science. 1982;217:1214–1222. - PubMed
-
- Yancey P. H. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 2005;208:2819–2830. - PubMed
-
- Schroer M. A. Zhai Y. Wieland D. F. Sahle C. J. Nase J. Paulus M. Tolan M. Winter R. Exploring the piezophilic behavior of natural cosolvent mixtures. Angew. Chem., Int. Ed. 2011;50:11413–11416. - PubMed
-
- Rani A. Venkatesu P. Changing relations between proteins and osmolytes: a choice of nature. Phys. Chem. Chem. Phys. 2018;20:20315–20333. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous