

Phylogenetic signatures reveal multilevel selection and fitness costs in SARS-CoV-2
- PMID: 39132669
- PMCID: PMC11316176
- DOI: 10.12688/wellcomeopenres.20704.2
Phylogenetic signatures reveal multilevel selection and fitness costs in SARS-CoV-2
Abstract
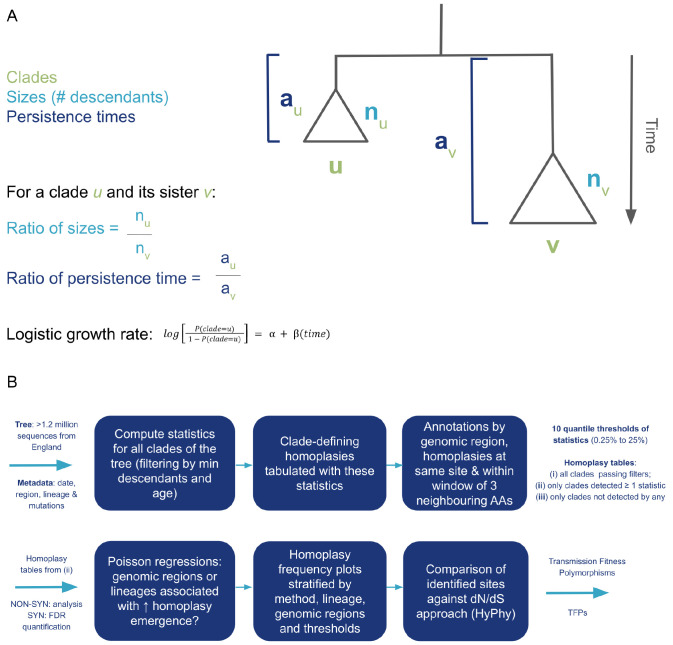
Background: Large-scale sequencing of SARS-CoV-2 has enabled the study of viral evolution during the COVID-19 pandemic. Some viral mutations may be advantageous to viral replication within hosts but detrimental to transmission, thus carrying a transient fitness advantage. By affecting the number of descendants, persistence times and growth rates of associated clades, these mutations generate localised imbalance in phylogenies. Quantifying these features in closely-related clades with and without recurring mutations can elucidate the tradeoffs between within-host replication and between-host transmission.
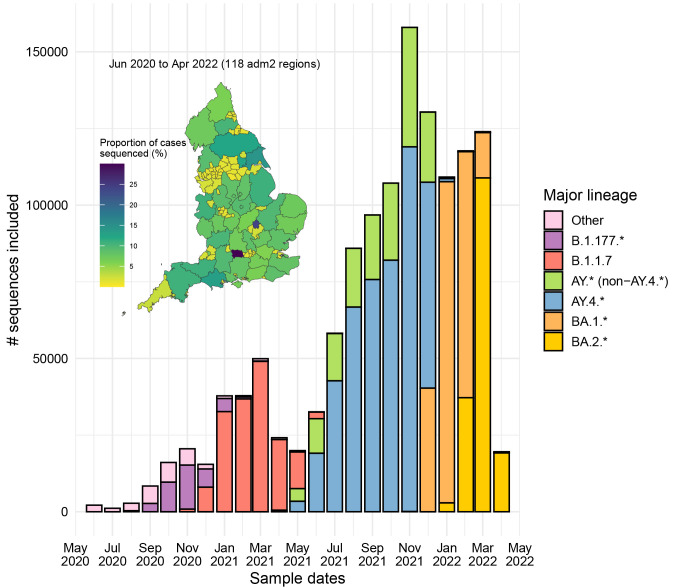
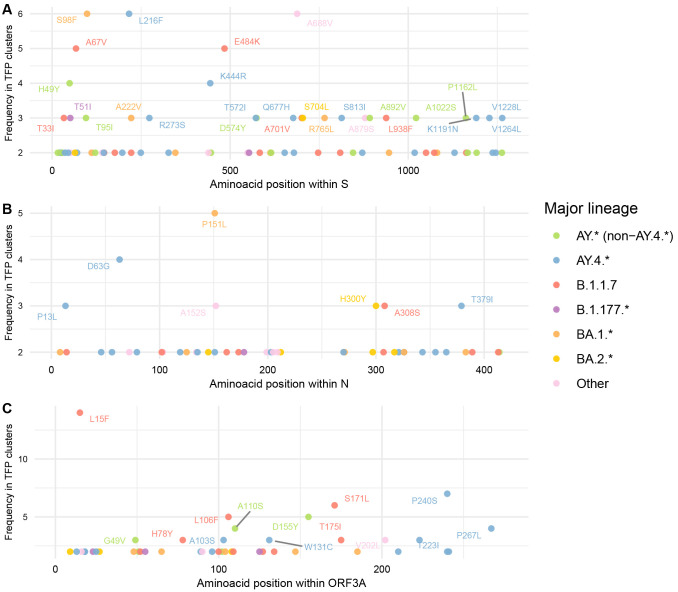
Methods: We implemented a novel phylogenetic clustering algorithm ( mlscluster, https://github.com/mrc-ide/mlscluster) to systematically explore time-scaled phylogenies for mutations under transient/multilevel selection. We applied this method to a SARS-CoV-2 time-calibrated phylogeny with >1.2 million sequences from England, and characterised these recurrent mutations that may influence transmission fitness across PANGO-lineages and genomic regions using Poisson regressions and summary statistics.
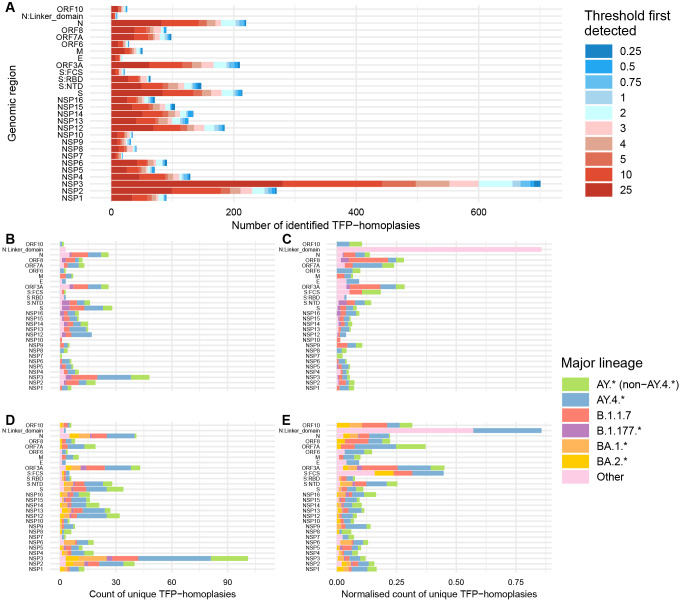
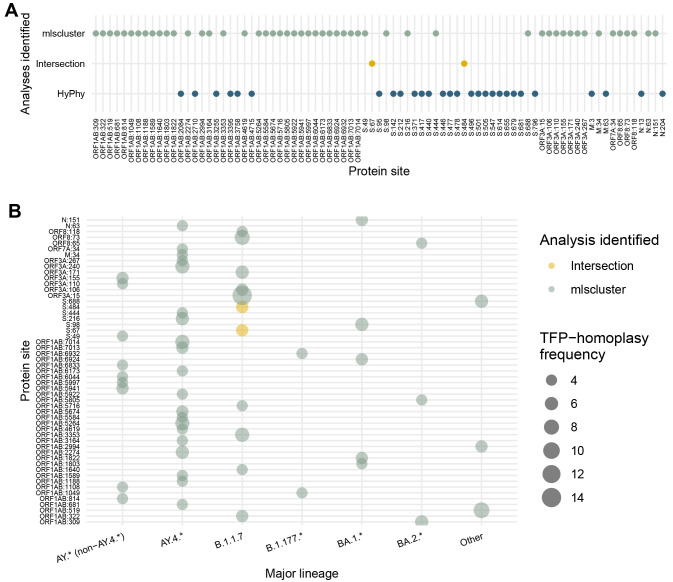
Results: We found no major differences across two epidemic stages (before and after Omicron), PANGO-lineages, and genomic regions. However, spike, nucleocapsid, and ORF3a were proportionally more enriched for transmission fitness polymorphisms (TFP)-homoplasies than other proteins. We provide a catalog of SARS-CoV-2 sites under multilevel selection, which can guide experimental investigations within and beyond the spike protein.
Conclusions: This study provides empirical evidence for the existence of important tradeoffs between within-host replication and between-host transmission shaping the fitness landscape of SARS-CoV-2. This method may be used as a fast and scalable means to shortlist large sequence databases for sites under putative multilevel selection which may warrant subsequent confirmatory analyses and experimental confirmation.
Keywords: Molecular evolution; SARS-CoV-2; genetic clustering; mutation; natural selection; phylogenetic analysis; transmission fitness; within-host evolution.
Plain language summary
Viral mutations can potentially carry a transient advantage, being simultaneously favourable for replication within hosts (e.g. by evading host immune responses) and deleterious to transmission (e.g. by having reduced cell binding). To identify such mutations, called transmission fitness polymorphisms (TFPs), we developed a clustering algorithm entitled mlscluster that computes clade-level statistics based on the number of descendants, persistence times, and growth rates of clades carrying a specific mutation in comparison with their immediate sisters without the mutation, which usually are different than expected in the presence of such TFPs. We then applied it to a representative SARS-CoV-2 time-scaled tree with >1 million whole-genome sequences from England. Our statistical analysis suggested approximately constant levels of transient selection across waves driven by very distinct variants. It also showed that genomic regions of known functional significance such as spike, nucleocapsid, and ORF3a were enriched for TFPs. This is the one of the first studies to characterise SARS-CoV-2 recurrent mutations potentially under multilevel selection, providing empirical evidence for the existence of important tradeoffs in selection between intrahost replication and inter-host transmission. Therefore, it provides target mutations for realistic coalescent-based modelling and laboratory-based investigations of their impacts and mechanisms of interaction with human cells.
Copyright: © 2024 Bonetti Franceschi V and Volz E.
Conflict of interest statement
No competing interests were disclosed.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
