

READv2: advanced and user-friendly detection of biological relatedness in archaeogenomics
- PMID: 39135108
- PMCID: PMC11318251
- DOI: 10.1186/s13059-024-03350-3
READv2: advanced and user-friendly detection of biological relatedness in archaeogenomics
Abstract
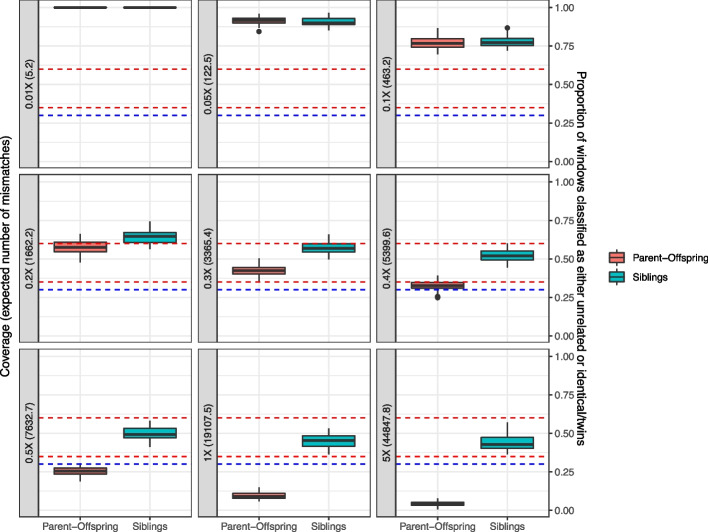
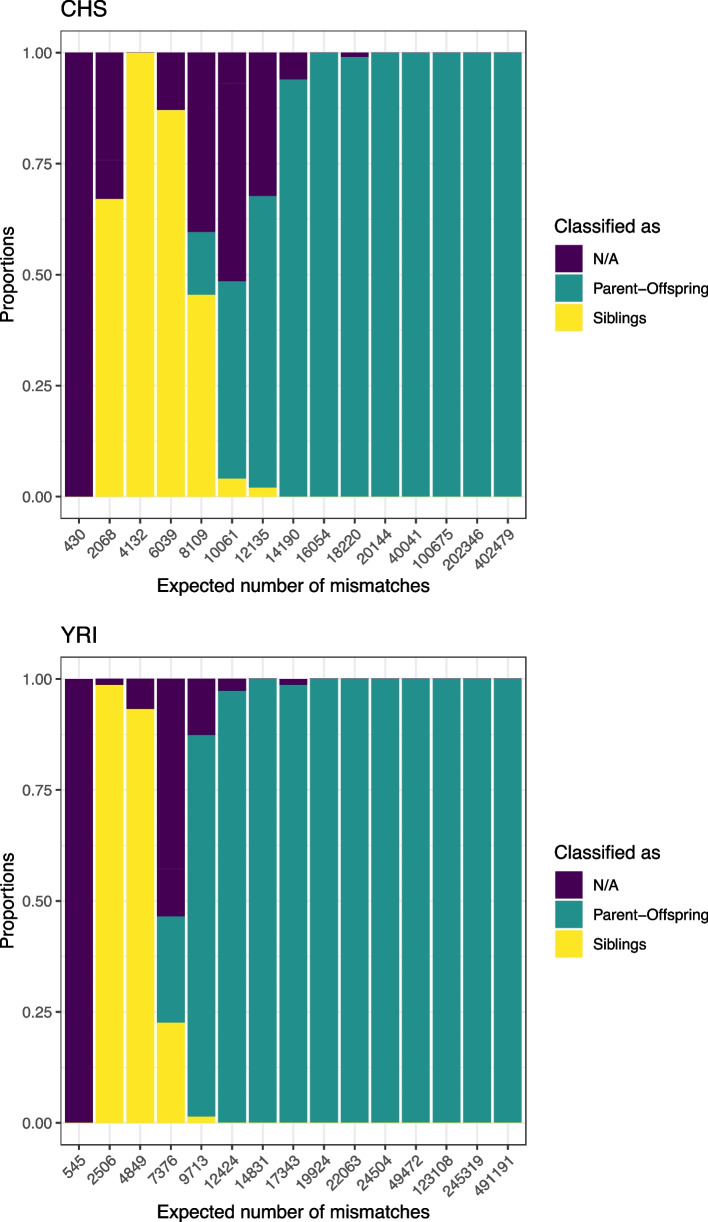
The advent of genome-wide ancient DNA analysis has revolutionized our understanding of prehistoric societies. However, studying biological relatedness in these groups requires tailored approaches due to the challenges of analyzing ancient DNA. READv2, an optimized Python3 implementation of the most widely used tool for this purpose, addresses these challenges while surpassing its predecessor in speed and accuracy. For sufficient amounts of data, it can classify up to third-degree relatedness and differentiate between the two types of first-degree relatedness, full siblings and parent-offspring. READv2 enables user-friendly, efficient, and nuanced analysis of biological relatedness, facilitating a deeper understanding of past social structures.
Keywords: Ancient DNA; Archaeogenomics; Kinship; Relatedness; Software.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures





References
-
- Orlando L, Allaby R, Skoglund P, Der Sarkissian C, Stockhammer PW, Ávila-Arcos MC, et al. Ancient DNA analysis. Nat Rev Methods Primers. 2021;1:1–26. 10.1038/s43586-020-00011-0.10.1038/s43586-020-00011-0 - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
