

Phosphorylation patterns in the AT1R C-terminal tail specify distinct downstream signaling pathways
- PMID: 39137246
- PMCID: PMC11443182
- DOI: 10.1126/scisignal.adk5736
Phosphorylation patterns in the AT1R C-terminal tail specify distinct downstream signaling pathways
Abstract
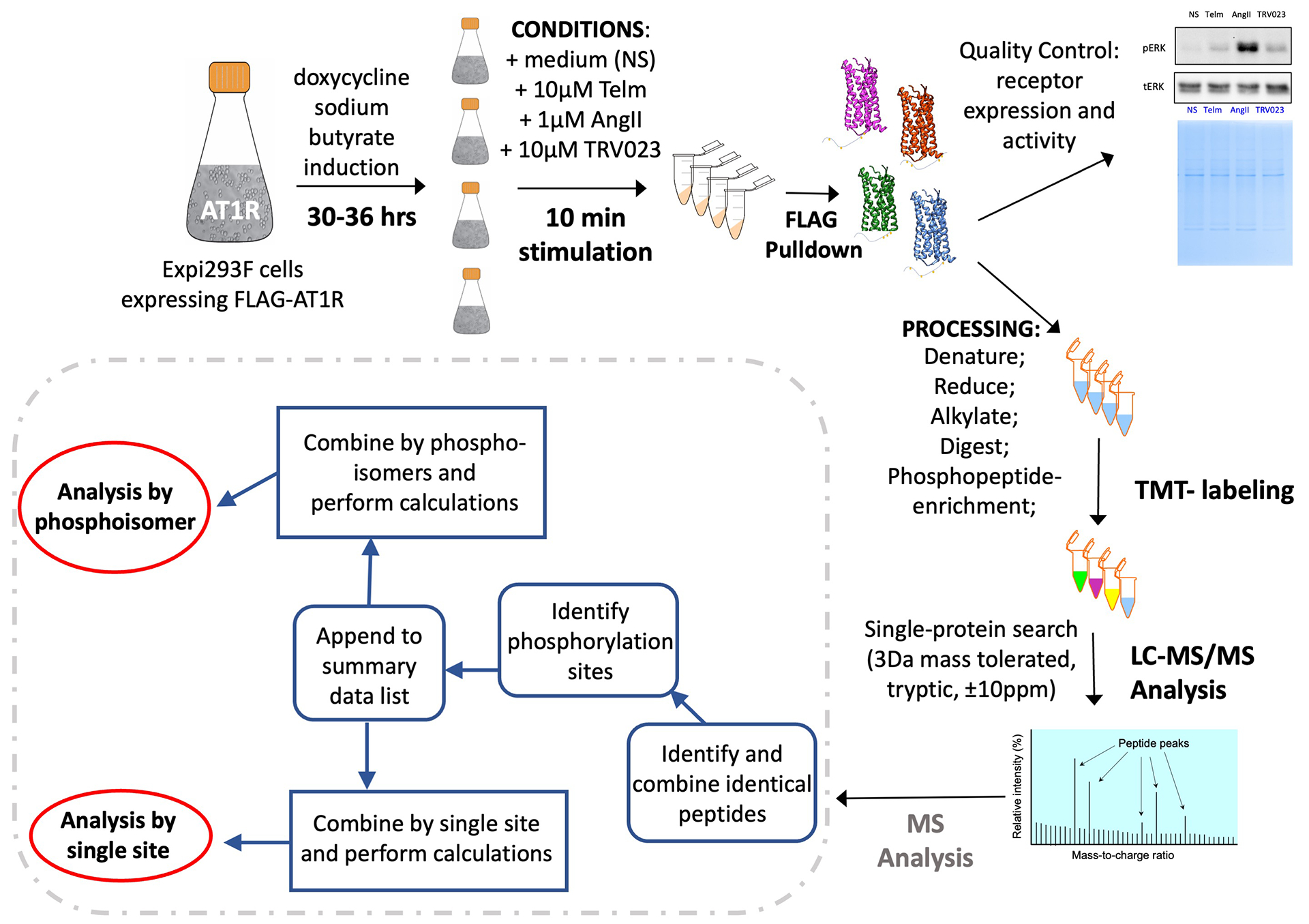
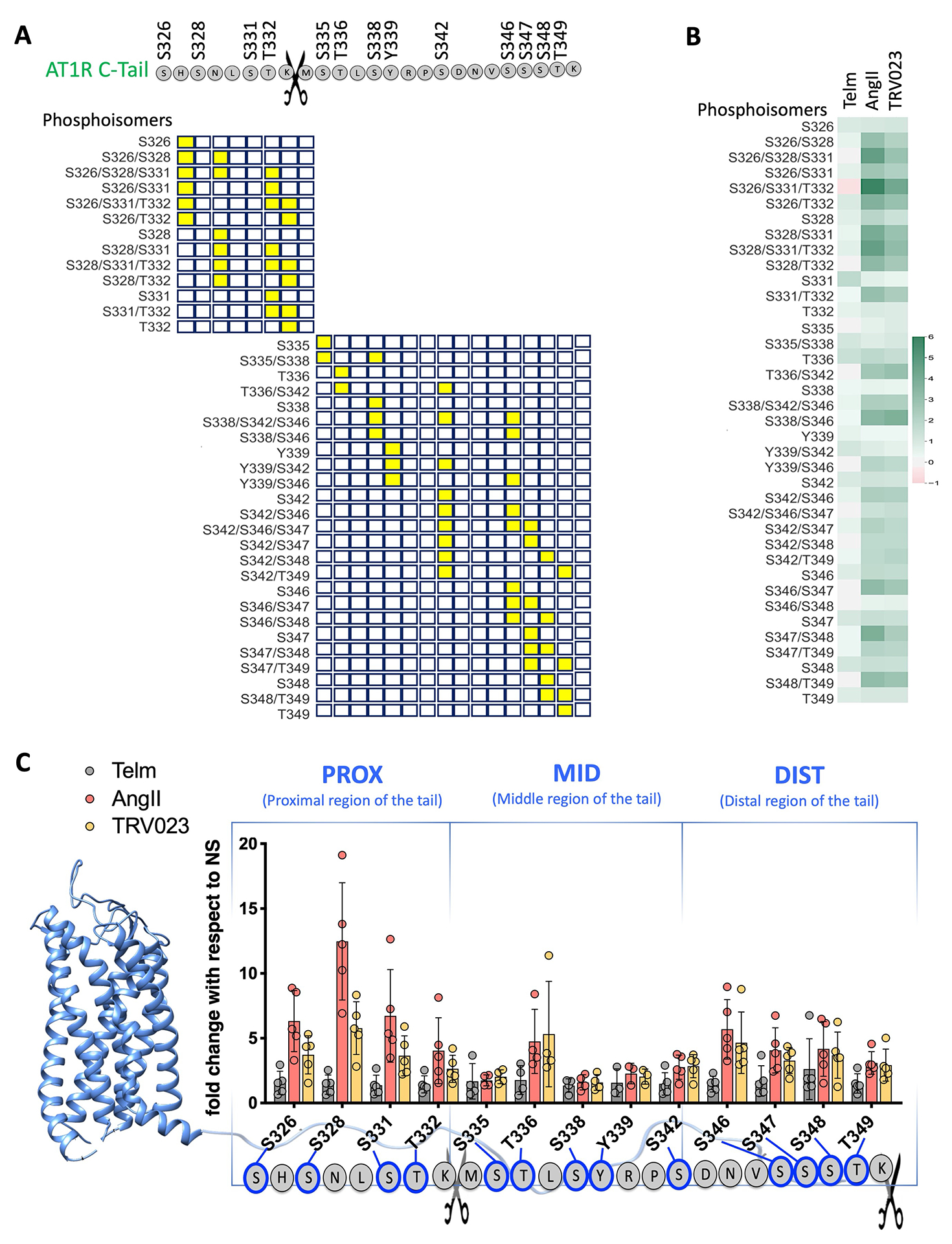
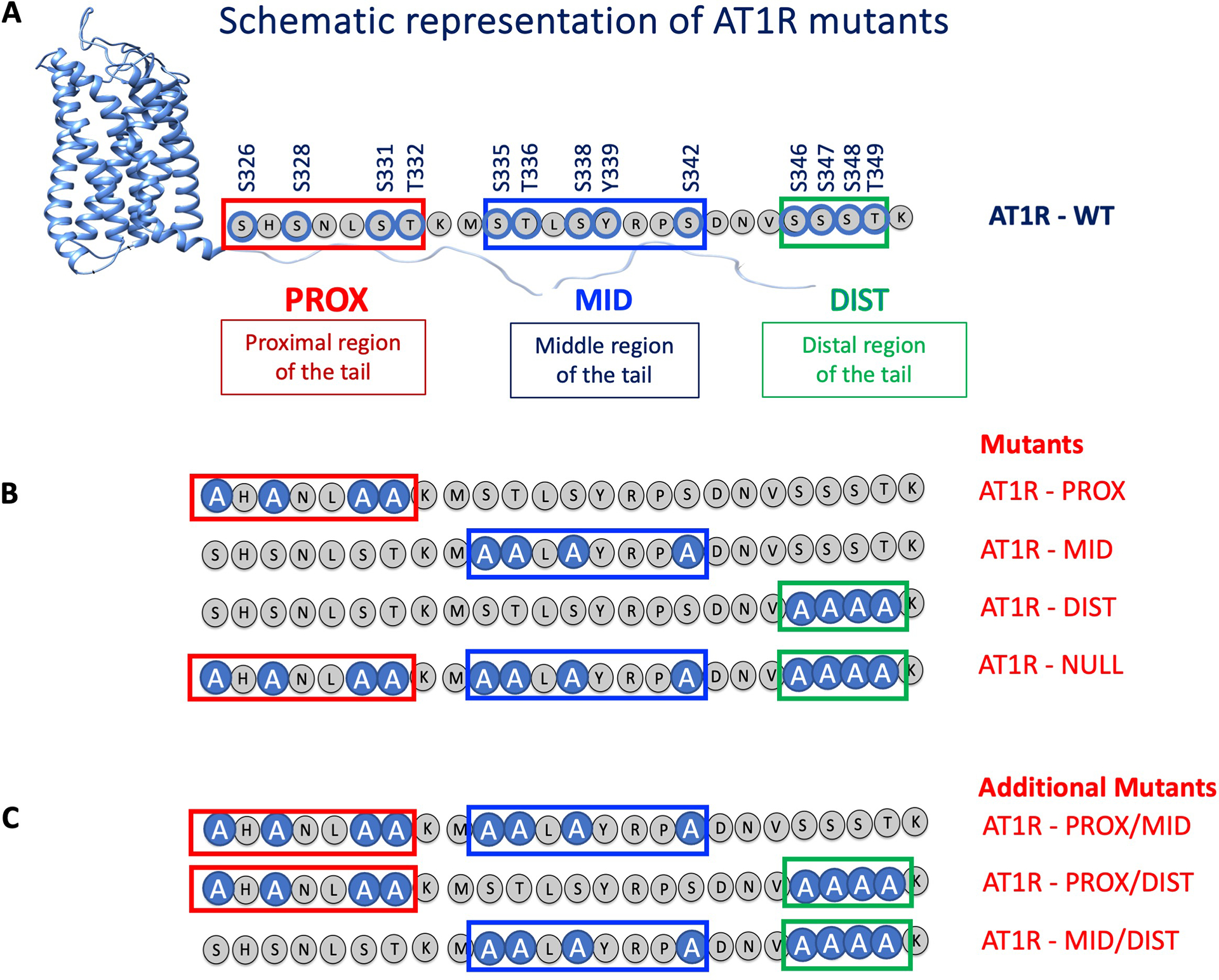
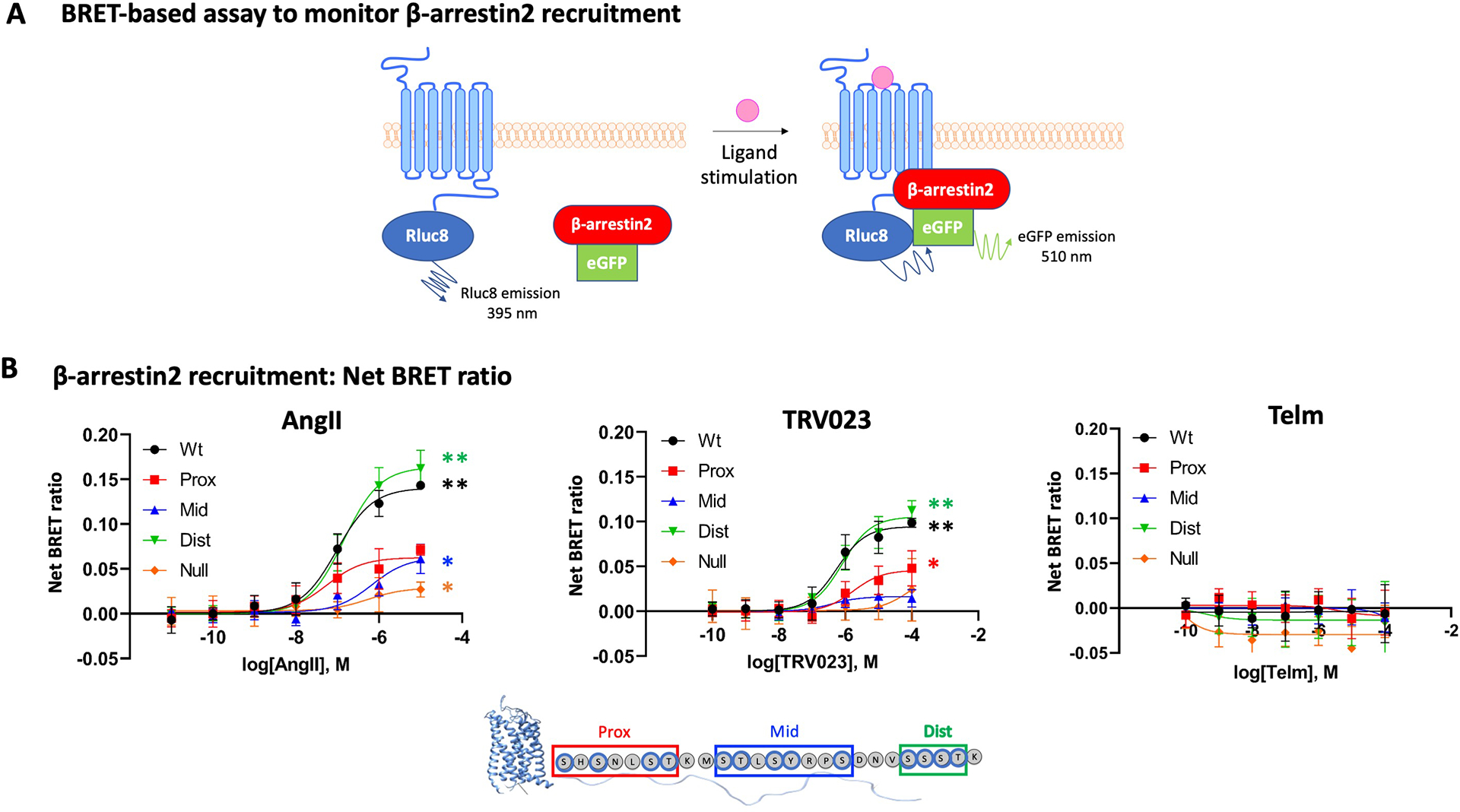
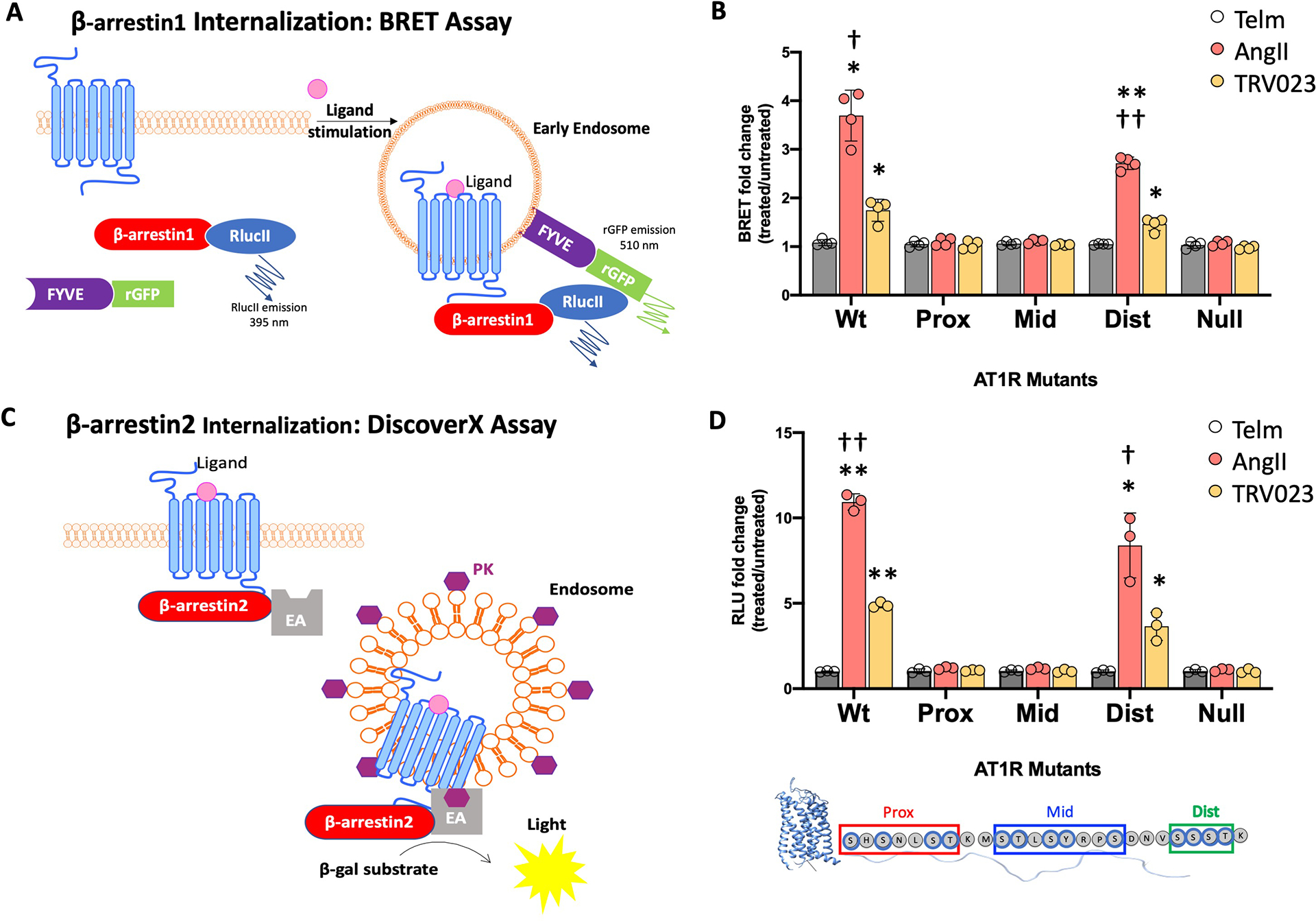
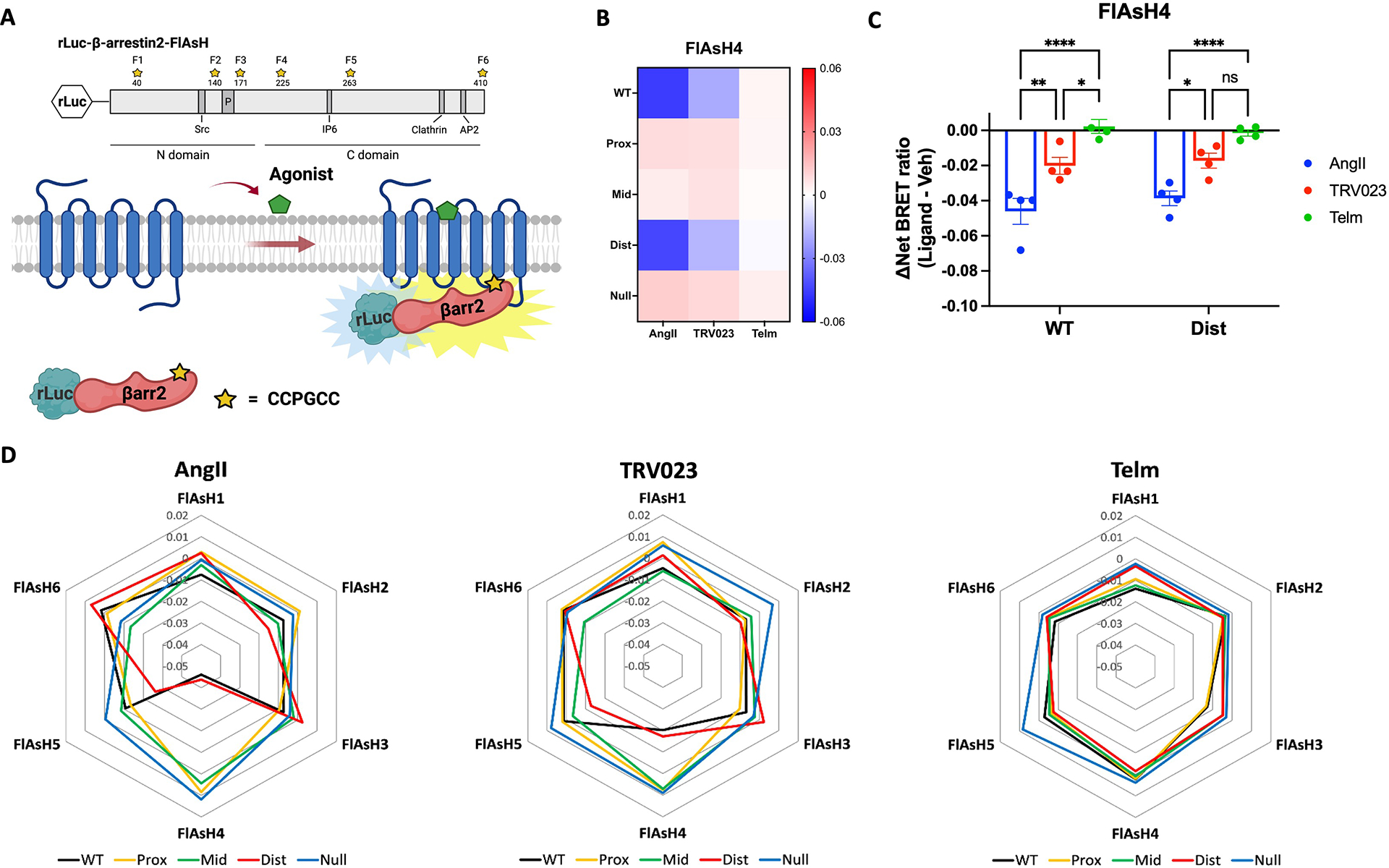
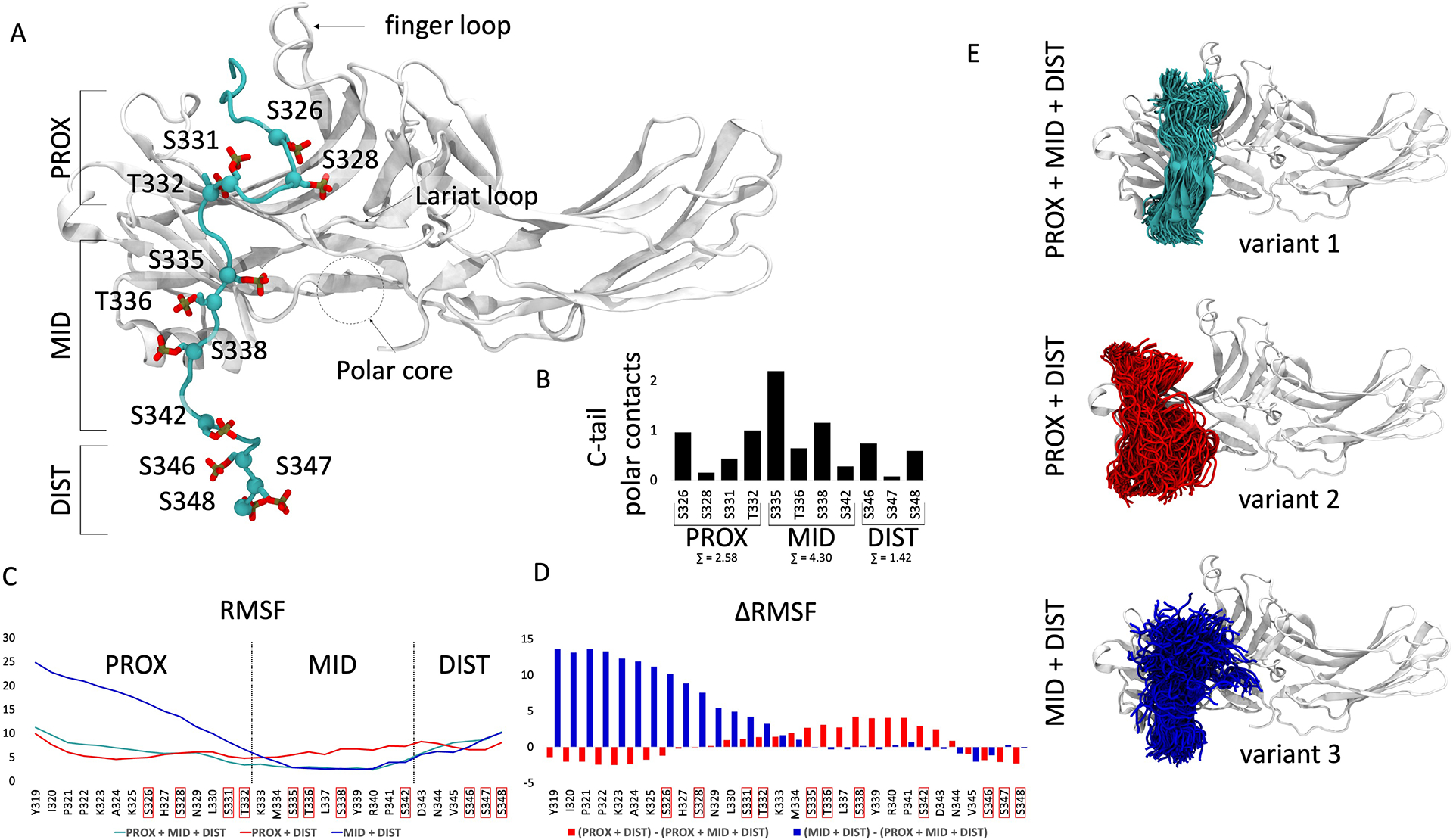
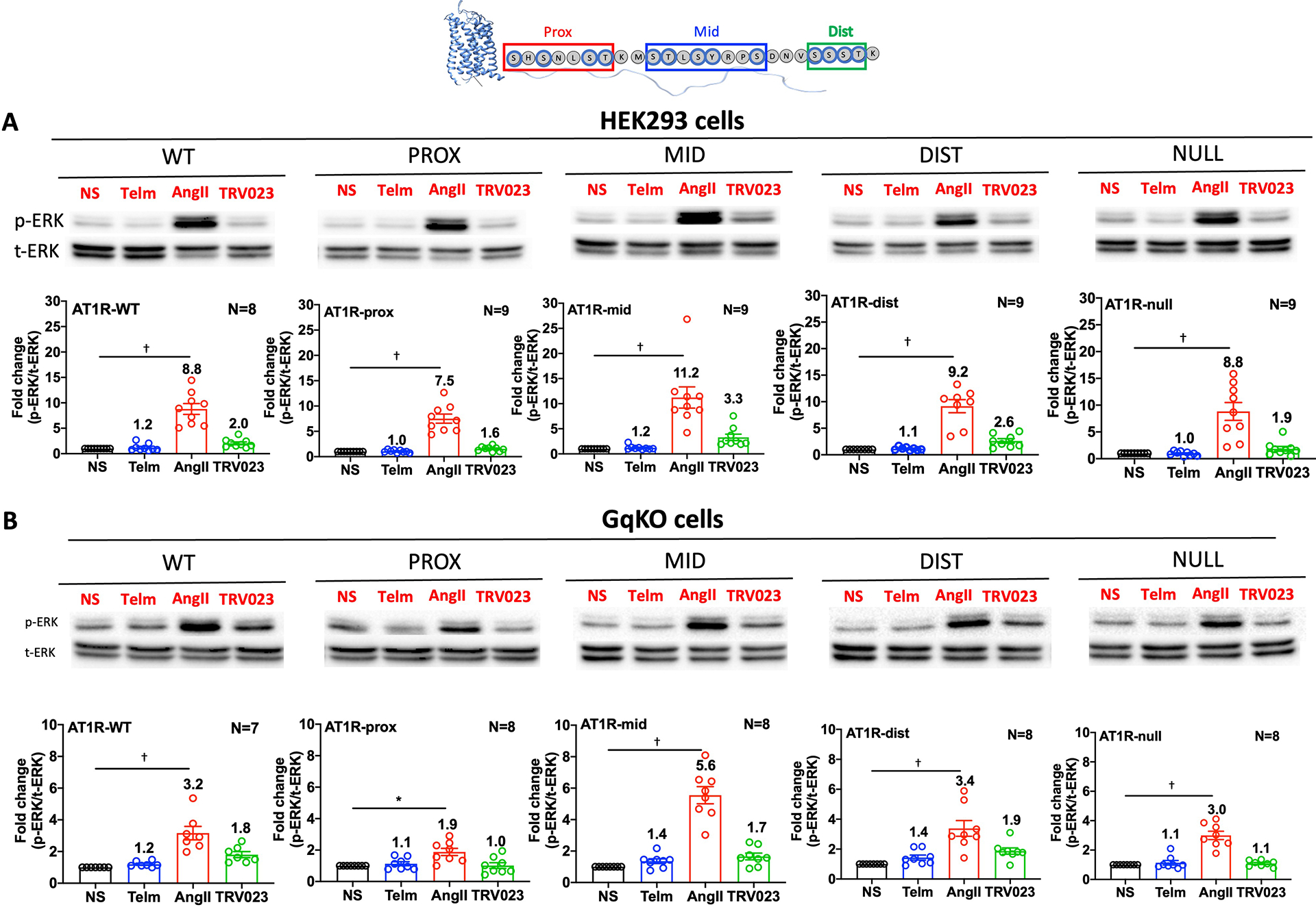
Different ligands stabilize specific conformations of the angiotensin II type 1 receptor (AT1R) that direct distinct signaling cascades mediated by heterotrimeric G proteins or β-arrestin. These different active conformations are thought to engage distinct intracellular transducers because of differential phosphorylation patterns in the receptor C-terminal tail (the "barcode" hypothesis). Here, we identified the AT1R barcodes for the endogenous agonist AngII, which stimulates both G protein activation and β-arrestin recruitment, and for a synthetic biased agonist that only stimulates β-arrestin recruitment. The endogenous and β-arrestin-biased agonists induced two different ensembles of phosphorylation sites along the C-terminal tail. The phosphorylation of eight serine and threonine residues in the proximal and middle portions of the tail was required for full β-arrestin functionality, whereas phosphorylation of the serine and threonine residues in the distal portion of the tail had little influence on β-arrestin function. Similarly, molecular dynamics simulations showed that the proximal and middle clusters of phosphorylated residues were critical for stable β-arrestin-receptor interactions. These findings demonstrate that ligands that stabilize different receptor conformations induce different phosphorylation clusters in the C-terminal tail as barcodes to evoke distinct receptor-transducer engagement, receptor trafficking, and signaling.
Conflict of interest statement
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
