

Targeting BCL2 with Venetoclax Enhances the Efficacy of the KRASG12D Inhibitor MRTX1133 in Pancreatic Cancer
- PMID: 39137400
- PMCID: PMC11532783
- DOI: 10.1158/0008-5472.CAN-23-3574
Targeting BCL2 with Venetoclax Enhances the Efficacy of the KRASG12D Inhibitor MRTX1133 in Pancreatic Cancer
Abstract
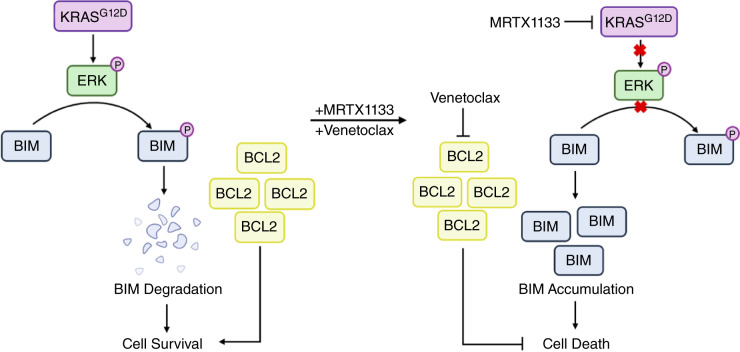
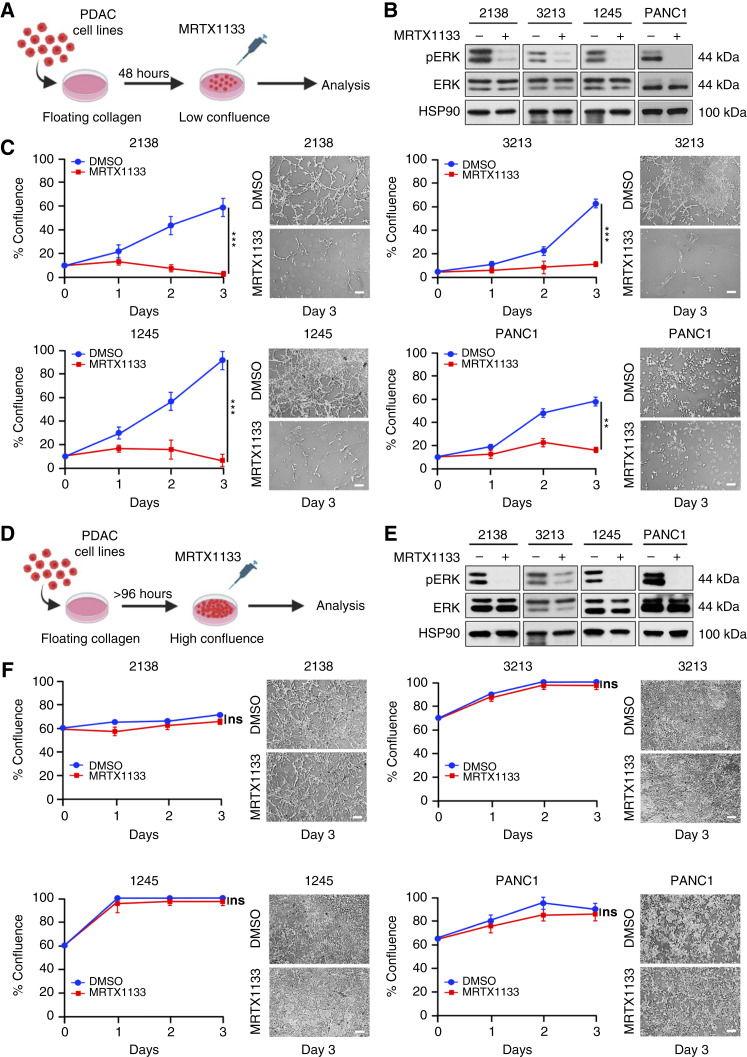
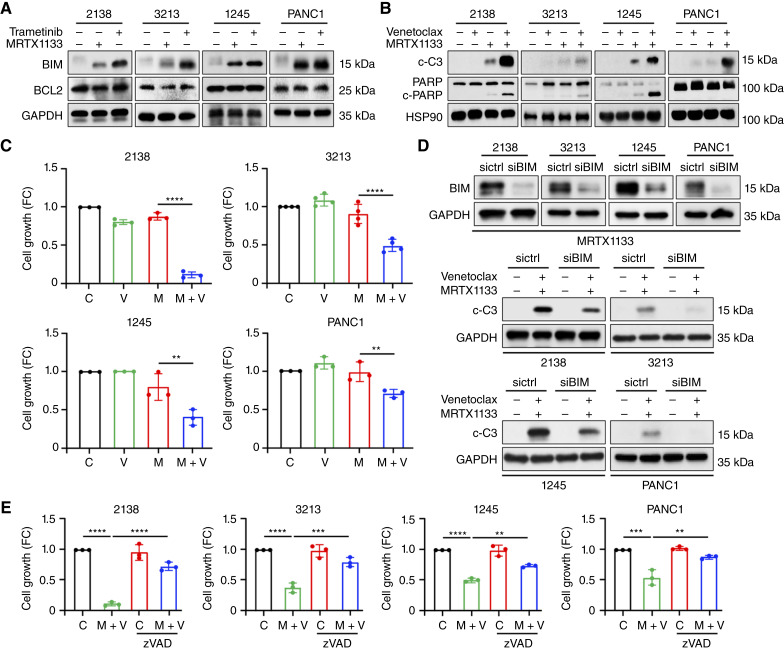
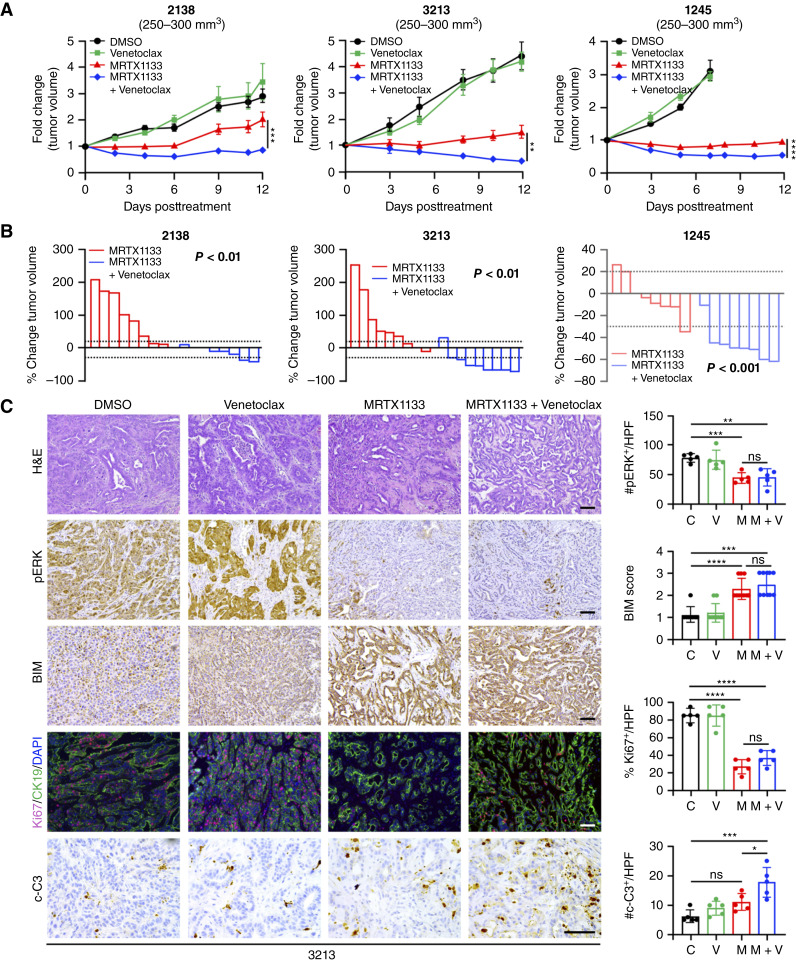
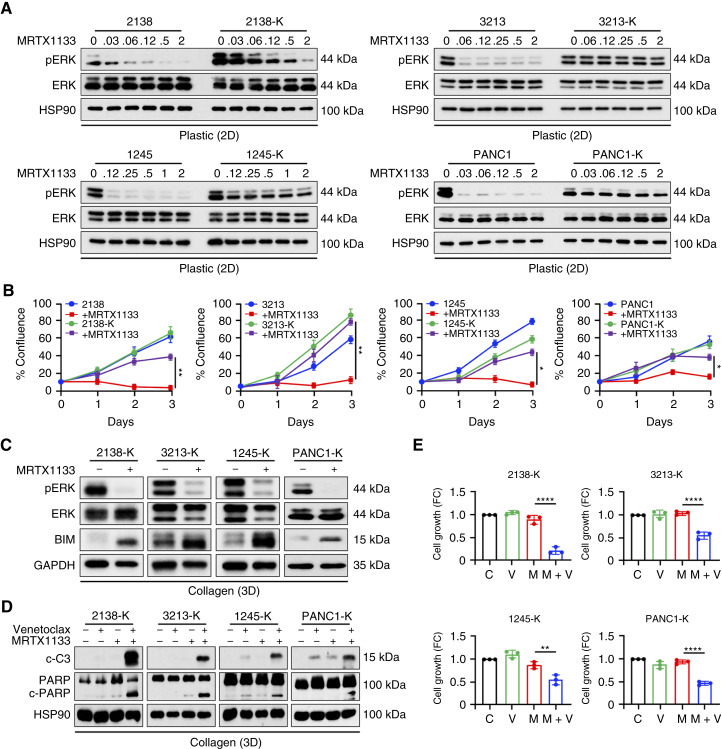
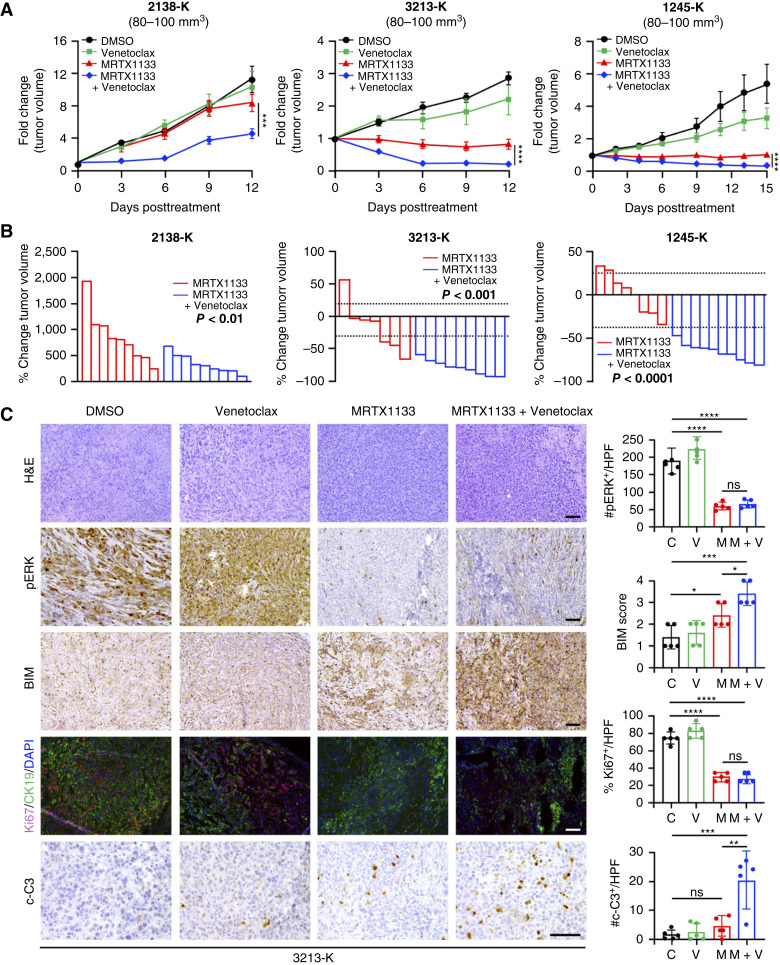
MRTX1133 is currently being evaluated in patients with pancreatic ductal adenocarcinoma (PDAC) tumors harboring a KRASG12D mutation. Combination strategies have the potential to enhance the efficacy of MRTX1133 to further promote cell death and tumor regression. In this study, we demonstrated that MRTX1133 increased the levels of the proapoptotic protein BIM in PDAC cells and conferred sensitivity to the FDA-approved BCL2 inhibitor venetoclax. Combined treatment with MRTX1133 and venetoclax resulted in cell death and growth suppression in 3D cultures. BIM was required for apoptosis induced by the combination treatment. Consistently, BIM was induced in tumors treated with MRTX1133, and venetoclax enhanced the efficacy of MRTX1133 in vivo. Venetoclax could also resensitize MRTX1133-resistant PDAC cells to MRTX1133 in 3D cultures, and tumors established from resistant cells responded to the combination of MRTX1133 and venetoclax. These results provide a rationale for the clinical testing of MRTX1133 and venetoclax in patients with PDAC. Significance: The combination of MRTX1133 and the FDA-approved drug venetoclax promotes cancer cell death and tumor regression in pancreatic ductal adenocarcinoma, providing rationale for testing venetoclax with KRASG12D inhibitors in patients with pancreatic cancer.
©2024 The Authors; Published by the American Association for Cancer Research.
Conflict of interest statement
No disclosures were reported.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
