

Macrophage-mediated myelin recycling fuels brain cancer malignancy
- PMID: 39137777
- PMCID: PMC11429458
- DOI: 10.1016/j.cell.2024.07.030
Macrophage-mediated myelin recycling fuels brain cancer malignancy
Abstract
Tumors growing in metabolically challenged environments, such as glioblastoma in the brain, are particularly reliant on crosstalk with their tumor microenvironment (TME) to satisfy their high energetic needs. To study the intricacies of this metabolic interplay, we interrogated the heterogeneity of the glioblastoma TME using single-cell and multi-omics analyses and identified metabolically rewired tumor-associated macrophage (TAM) subpopulations with pro-tumorigenic properties. These TAM subsets, termed lipid-laden macrophages (LLMs) to reflect their cholesterol accumulation, are epigenetically rewired, display immunosuppressive features, and are enriched in the aggressive mesenchymal glioblastoma subtype. Engulfment of cholesterol-rich myelin debris endows subsets of TAMs to acquire an LLM phenotype. Subsequently, LLMs directly transfer myelin-derived lipids to cancer cells in an LXR/Abca1-dependent manner, thereby fueling the heightened metabolic demands of mesenchymal glioblastoma. Our work provides an in-depth understanding of the immune-metabolic interplay during glioblastoma progression, thereby laying a framework to unveil targetable metabolic vulnerabilities in glioblastoma.
Keywords: cancer immunity; cholesterol; glioblastoma; lipid metabolism; macrophages; myelin recycling; tumor microenvironment.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests L.A., D.J.K., A.K., and J.E. are inventors on European patents P091147NL and P099572EP describing relevant claims and their therapeutic potential.
Figures



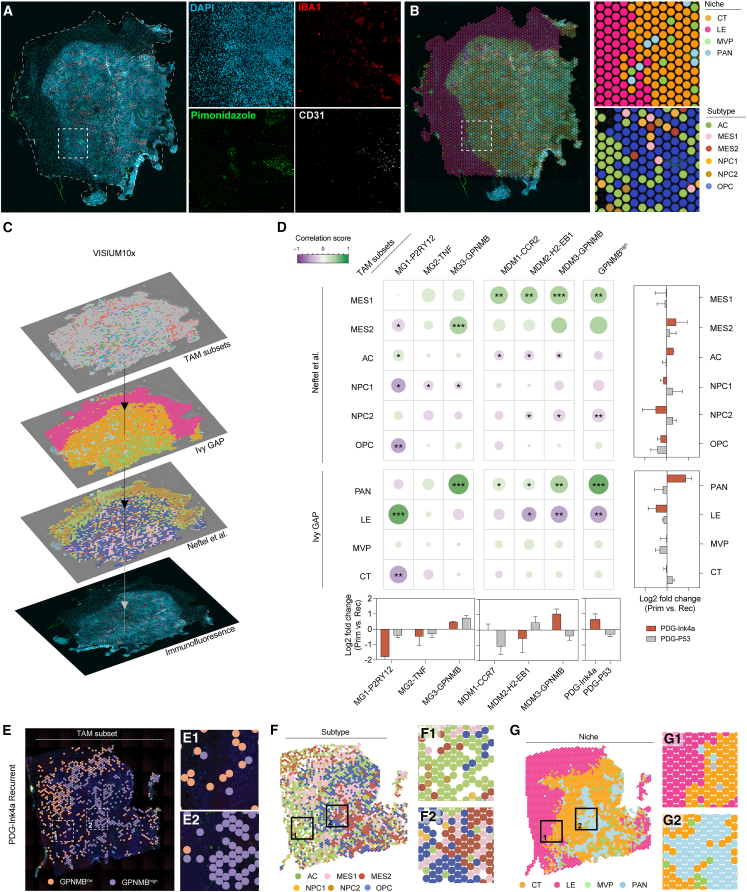




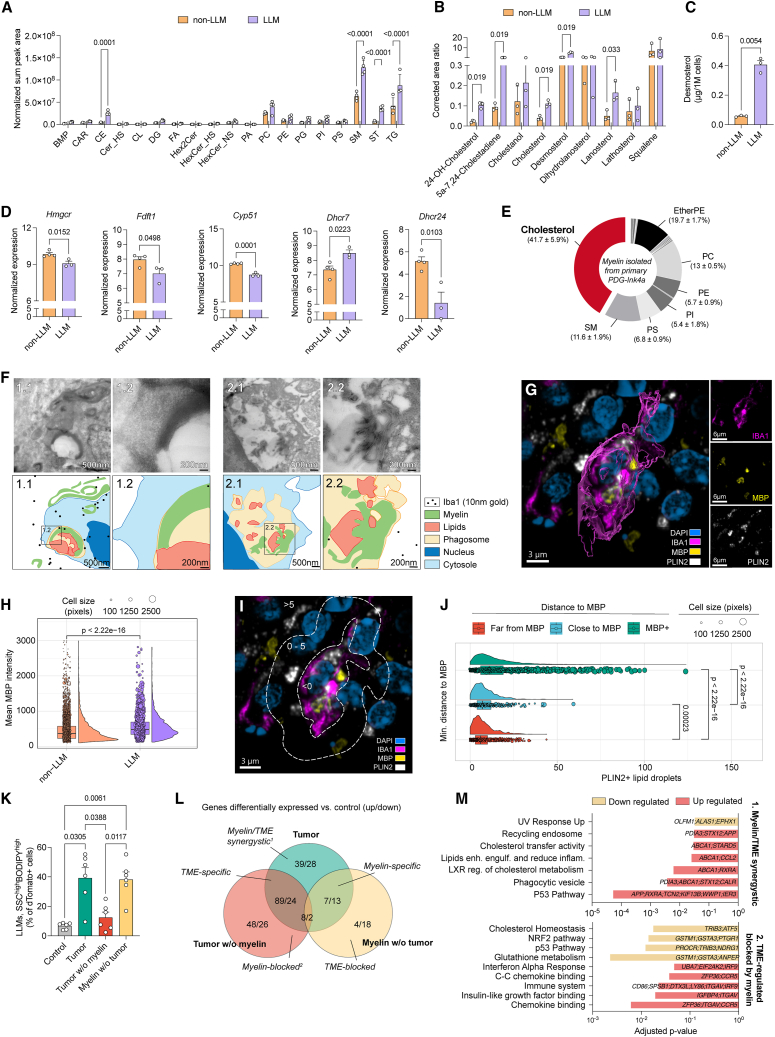







References
-
- Klemm F., Maas R.R., Bowman R.L., Kornete M., Soukup K., Nassiri S., Brouland J.-P., Iacobuzio-Donahue C.A., Brennan C., Tabar V., et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell. 2020;181:1643–1660.e17. doi: 10.1016/j.cell.2020.05.007. - DOI - PMC - PubMed
-
- Friebel E., Kapolou K., Unger S., Núñez N.G., Utz S., Rushing E.J., Regli L., Weller M., Greter M., Tugues S., et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell. 2020;181:1626–1642.e20. doi: 10.1016/j.cell.2020.04.055. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
