

Inhibition of glycosphingolipid synthesis with eliglustat in combination with immune checkpoint inhibitors in advanced cancers: preclinical evidence and phase I clinical trial
- PMID: 39138212
- PMCID: PMC11322526
- DOI: 10.1038/s41467-024-51495-3
Inhibition of glycosphingolipid synthesis with eliglustat in combination with immune checkpoint inhibitors in advanced cancers: preclinical evidence and phase I clinical trial
Abstract
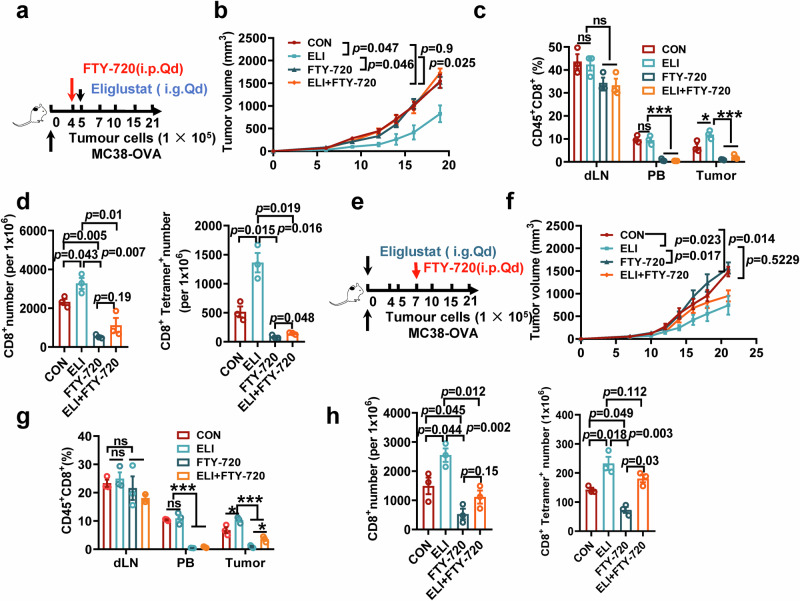
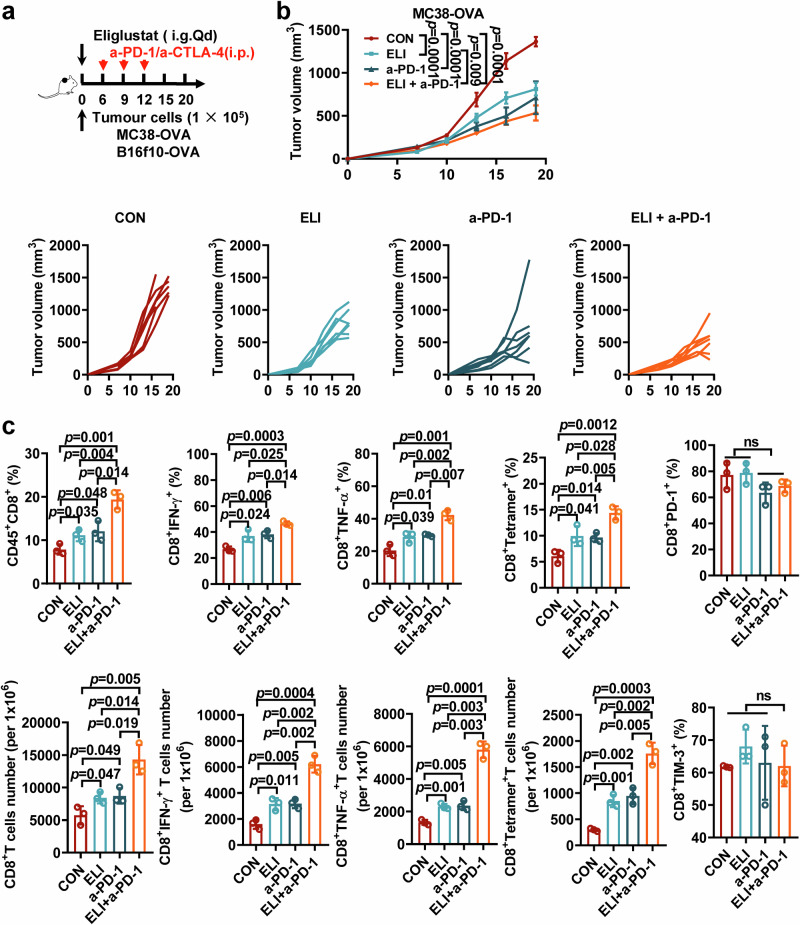
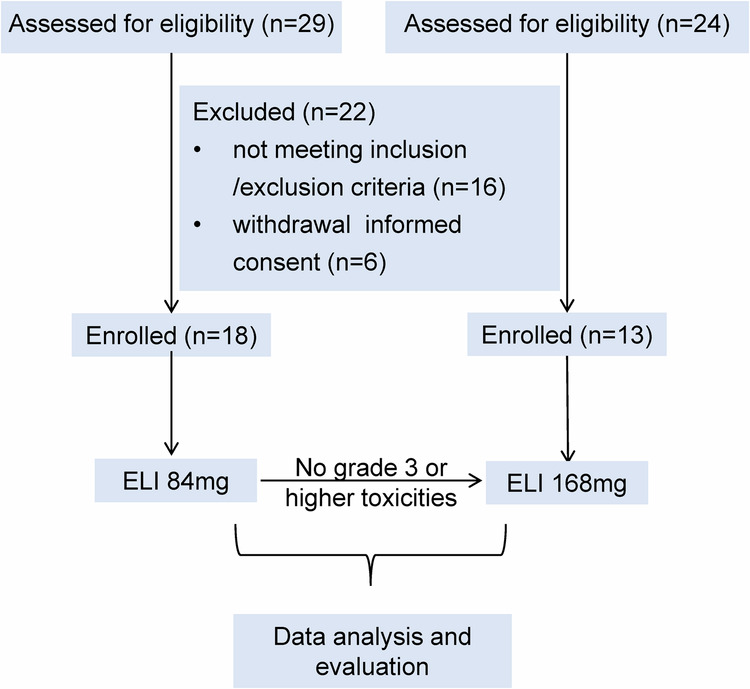
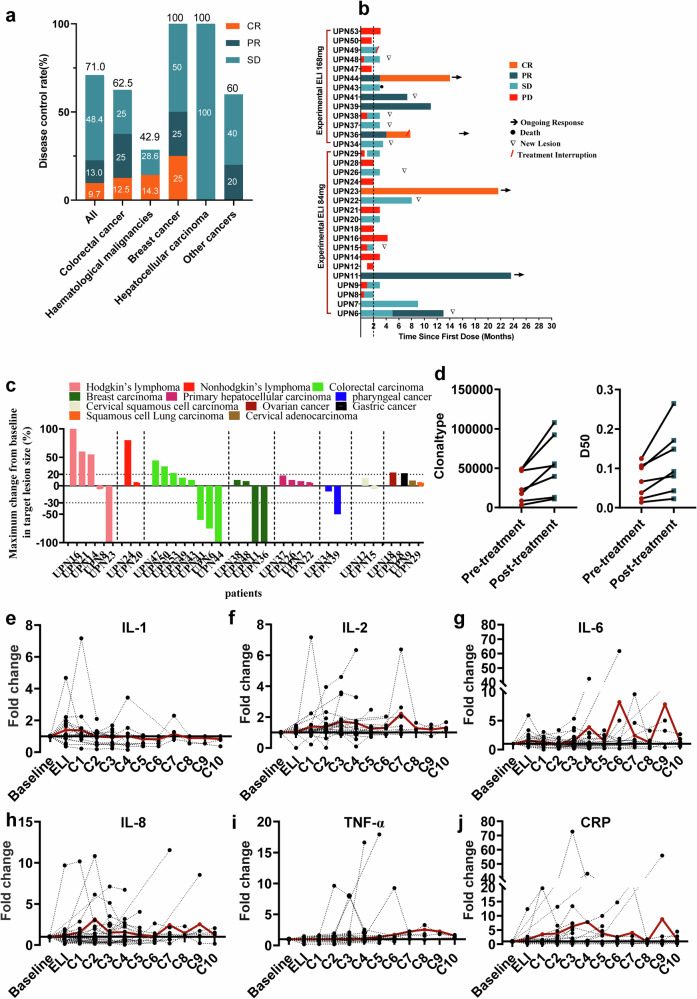
Glycosphingolipids (GSLs) are abundantly expressed in cancer cells. The effects of GSL-targeted immunotherapies are not fully understood. Here, we show that the inhibition of GSL synthesis with the UDP-glucose ceramide glucosyltransferase inhibitor eliglustat can increase the exposure of the major histocompatibility complex (MHC) and tumour antigen peptides, enhancing the antitumour response of CD8+ T cells in a range of tumour models. We therefore conducted a proof-of-concept phase I trial on the combination of eliglustat and an anti-PD-1 antibody for the treatment of advanced cancers (NCT04944888). The primary endpoints were safety and feasibility, and the secondary endpoint was antitumor activity. All prespecified endpoints were met. Among the 31 enrolled patients, only 1 patient experienced a grade 3 adverse event (AE), and no grade 4 AEs were observed. The objective response rate was 22.6% and the disease control rate reached 71%. Of the 8 patients with proficient mismatch repair/microsatellite stable (pMMR/MSS) colorectal cancer, one achieved complete response and two each had partial response and stable disease. In summary, inhibiting the synthesis of GSLs might represent an effective immunotherapy approach.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
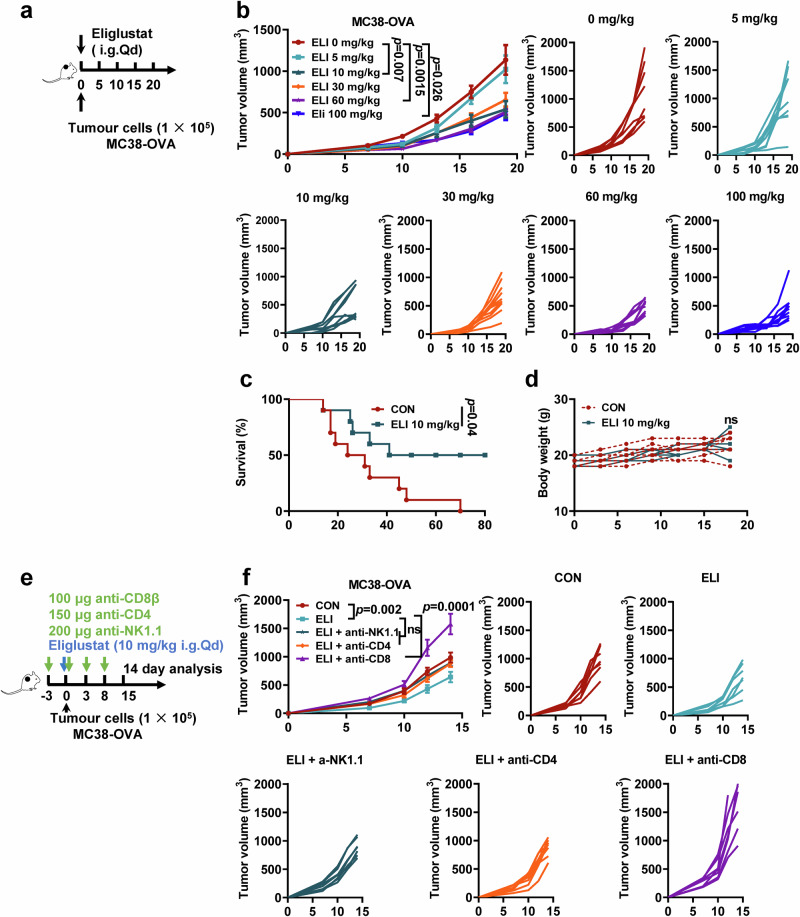
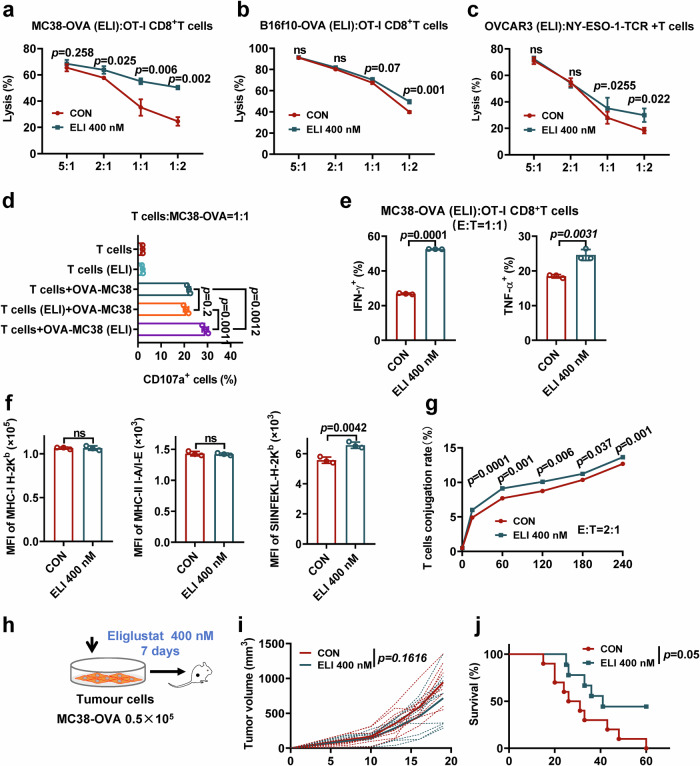
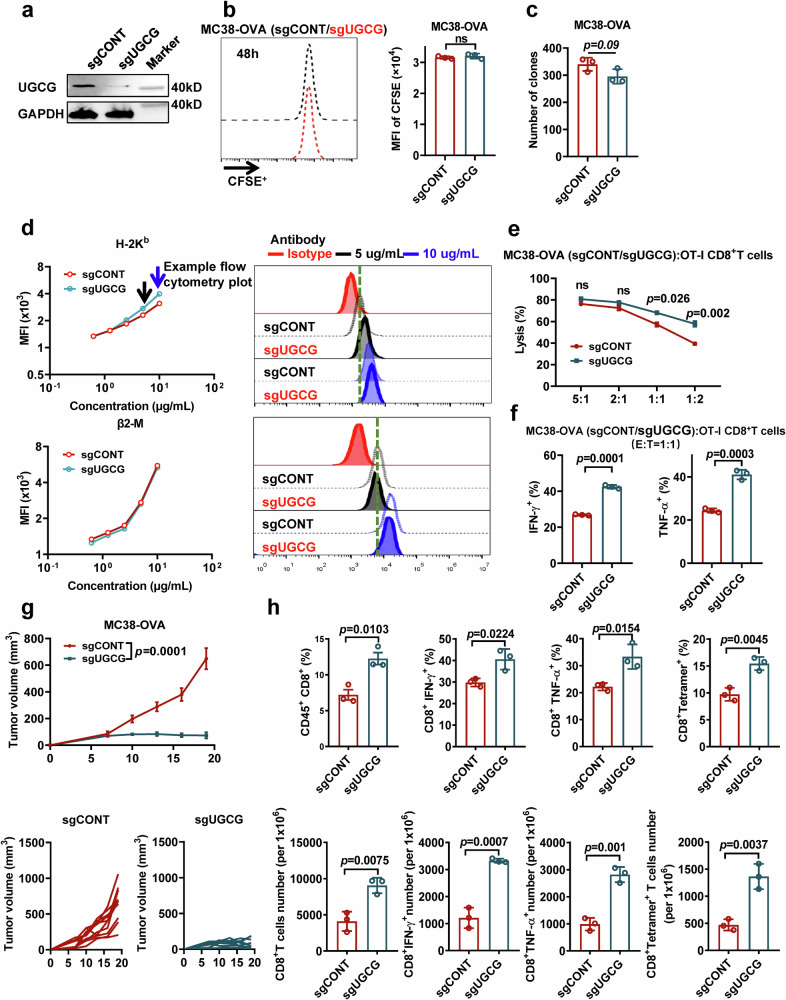
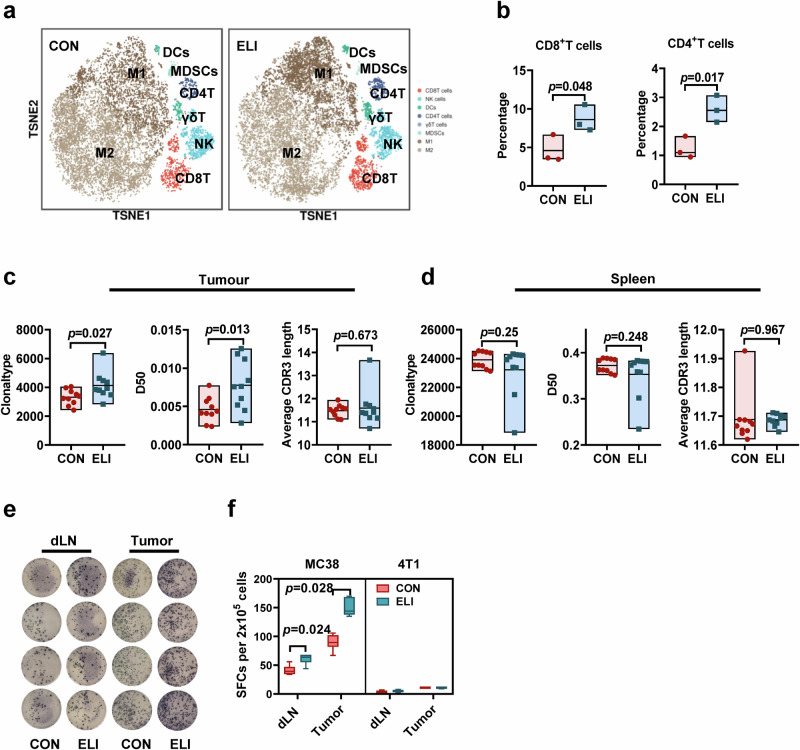
References
Publication types
MeSH terms
Substances
Grants and funding
- 82150108/National Natural Science Foundation of China (National Science Foundation of China)
- 31991171/National Natural Science Foundation of China (National Science Foundation of China)
- 82341208/National Natural Science Foundation of China (National Science Foundation of China)
- 82370226/National Natural Science Foundation of China (National Science Foundation of China)
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
