

Pathogenesis-driven treatment of primary pulmonary alveolar proteinosis
- PMID: 39142709
- PMCID: PMC11322829
- DOI: 10.1183/16000617.0064-2024
Pathogenesis-driven treatment of primary pulmonary alveolar proteinosis
Abstract
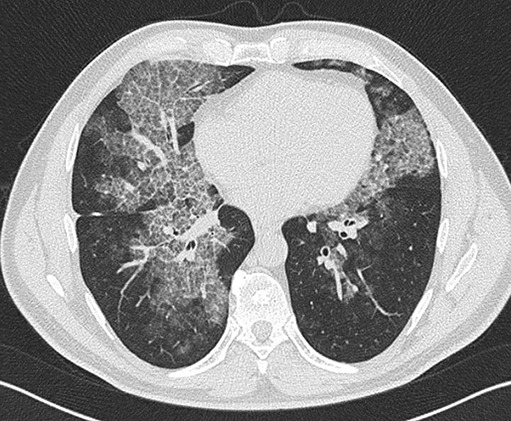
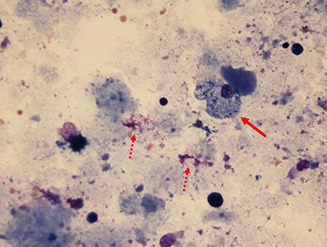
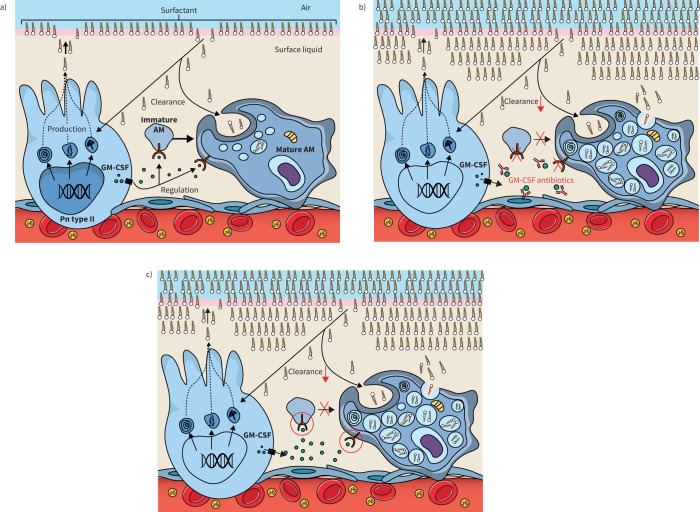
Pulmonary alveolar proteinosis (PAP) is a syndrome that results from the accumulation of lipoproteinaceous material in the alveolar space. According to the underlying pathogenetic mechanisms, three different forms have been identified, namely primary, secondary and congenital. Primary PAP is caused by disruption of granulocyte-macrophage colony-stimulating factor (GM-CSF) signalling due to the presence of neutralising autoantibodies (autoimmune PAP) or GM-CSF receptor genetic defects (hereditary PAP), which results in dysfunctional alveolar macrophages with reduced phagocytic clearance of particles, cholesterol and surfactant. The serum level of GM-CSF autoantibody is the only disease-specific biomarker of autoimmune PAP, although it does not correlate with disease severity. In PAP patients with normal serum GM-CSF autoantibody levels, elevated serum GM-CSF levels is highly suspicious for hereditary PAP. Several biomarkers have been correlated with disease severity, although they are not specific for PAP. These include lactate dehydrogenase, cytokeratin 19 fragment 21.1, carcinoembryonic antigen, neuron-specific enolase, surfactant proteins, Krebs von Lungen 6, chitinase-3-like protein 1 and monocyte chemotactic proteins. Finally, increased awareness of the disease mechanisms has led to the development of pathogenesis-based treatments, such as GM-CSF augmentation and cholesterol-targeting therapies.
Copyright ©The authors 2024.
Conflict of interest statement
Conflict of interest: F. Bonella reports consultancy fees from Boehringer Ingelheim, Sanofi, BMS and CSL-Behring, payment or honoraria for lectures, presentations, manuscript writing or educational events from Boehringer Ingelheim and Sanofi, support for attending meetings from Boehringer Ingelheim, Astra Zeneca and Atyr, and participation on a data safety monitoring board or advisory board with Boehringer Ingelheim, Sanofi and BMS. I. Campo reports consultancy fees from Partner Therapeutics, and participation on a data safety monitoring board or advisory board with Savara. All other authors have nothing to disclose.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials