

Branched-chain amino acid transaminase 1 confers EGFR-TKI resistance through epigenetic glycolytic activation
- PMID: 39143065
- PMCID: PMC11324870
- DOI: 10.1038/s41392-024-01928-8
Branched-chain amino acid transaminase 1 confers EGFR-TKI resistance through epigenetic glycolytic activation
Abstract
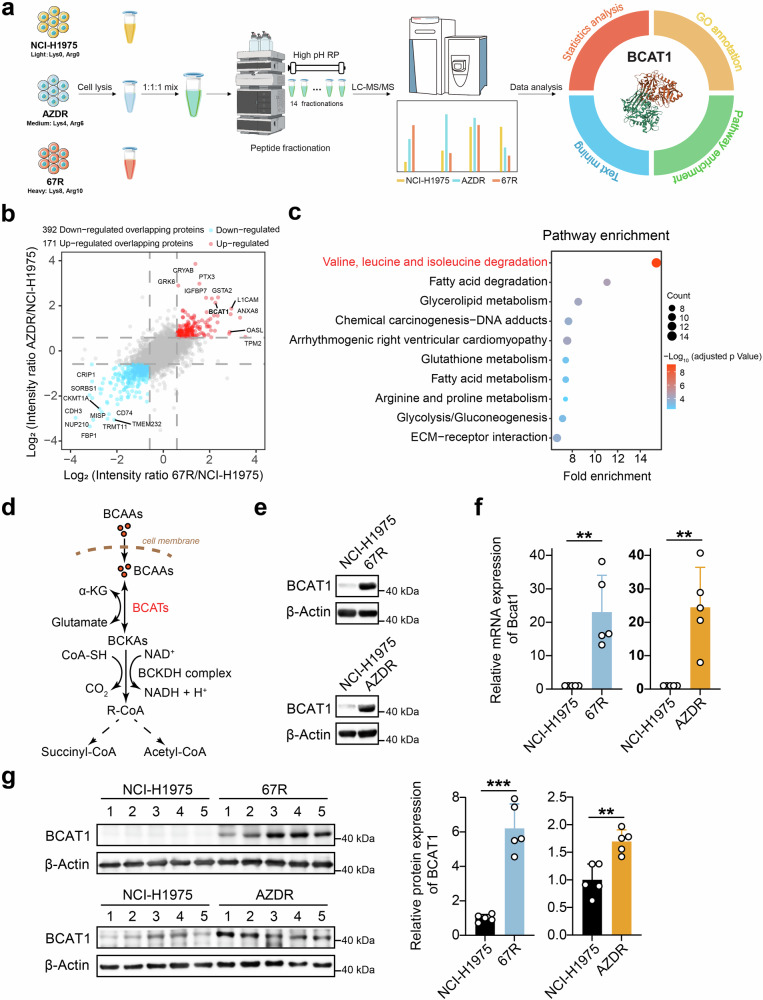
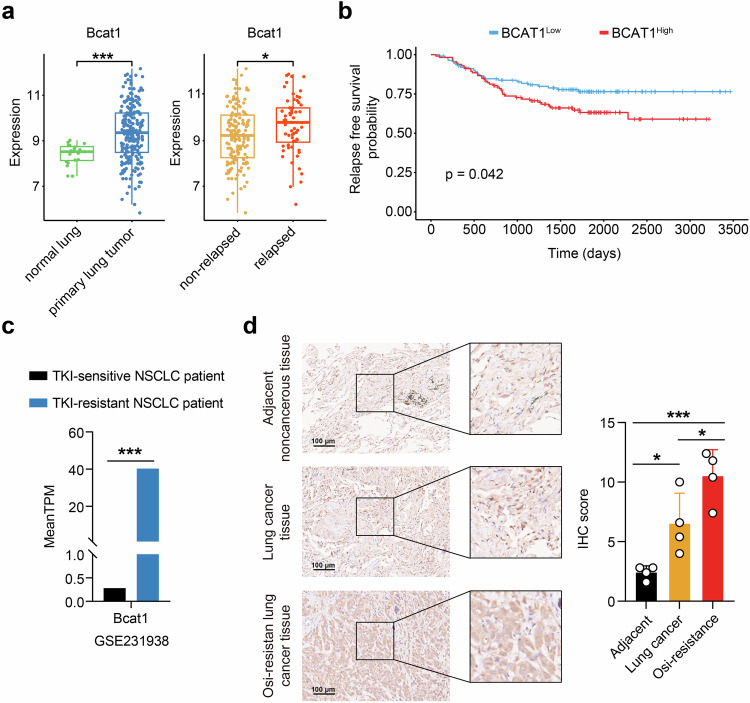
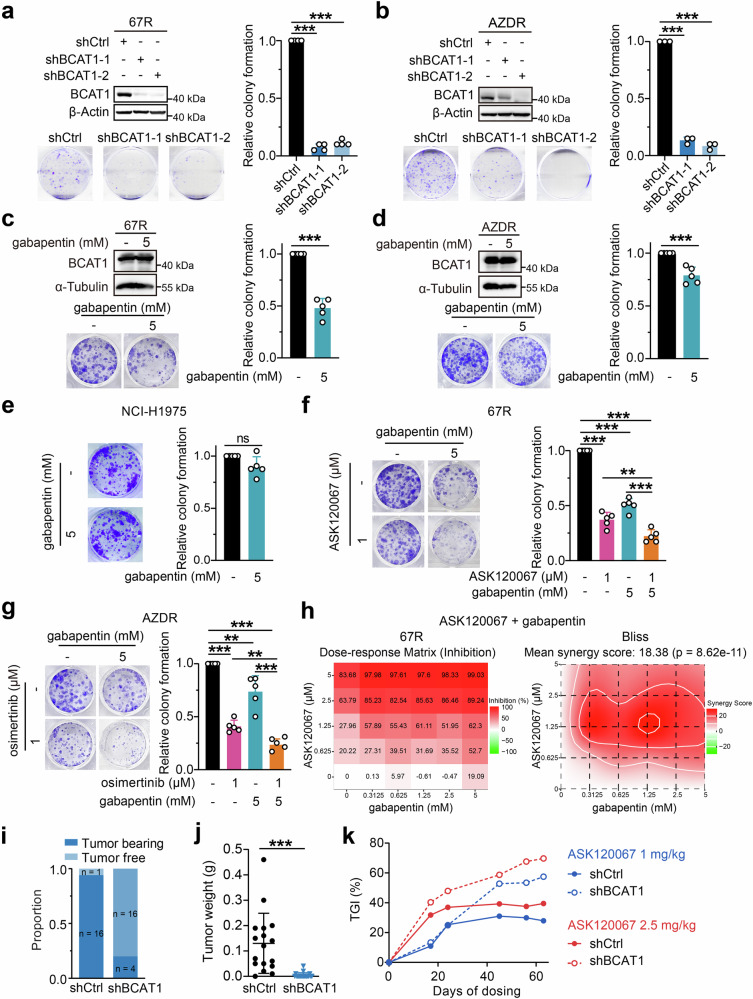
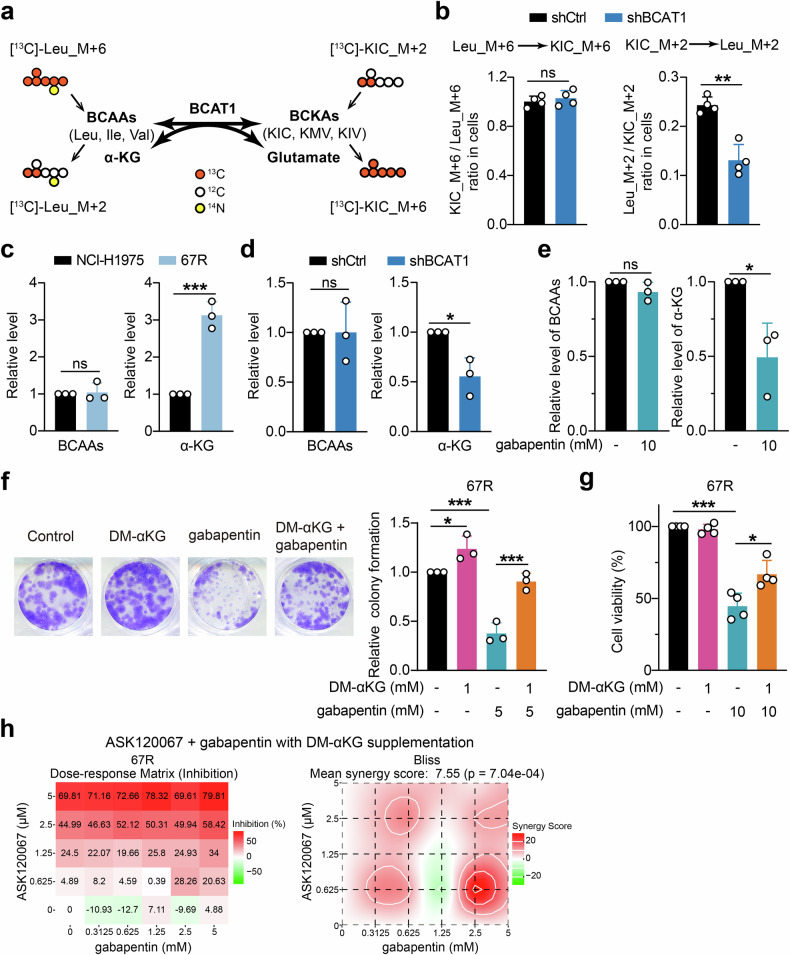
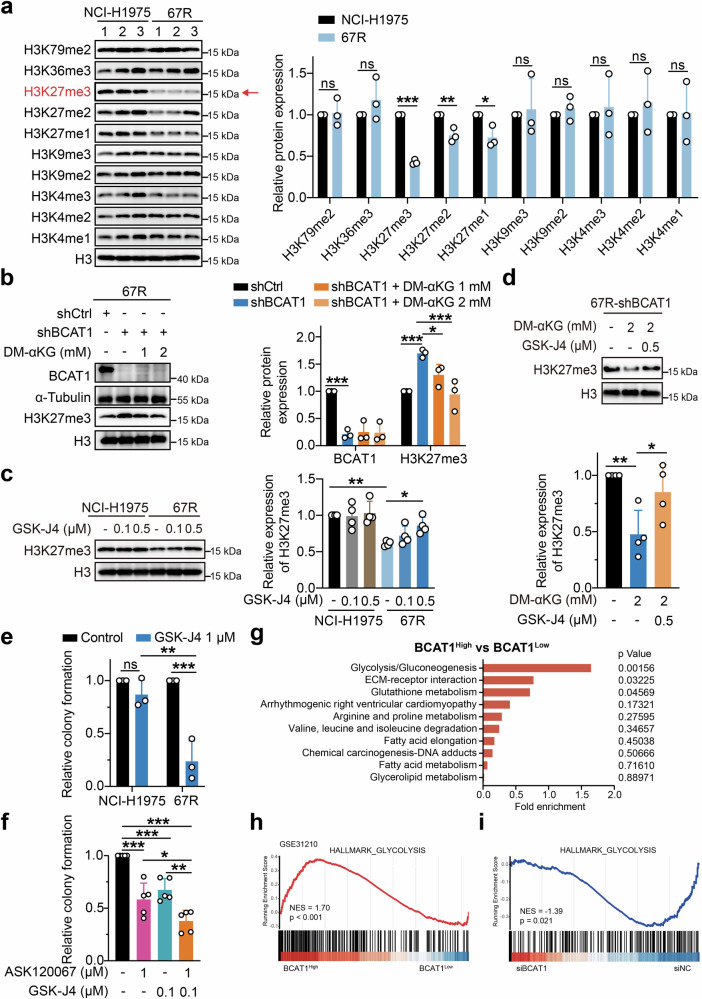
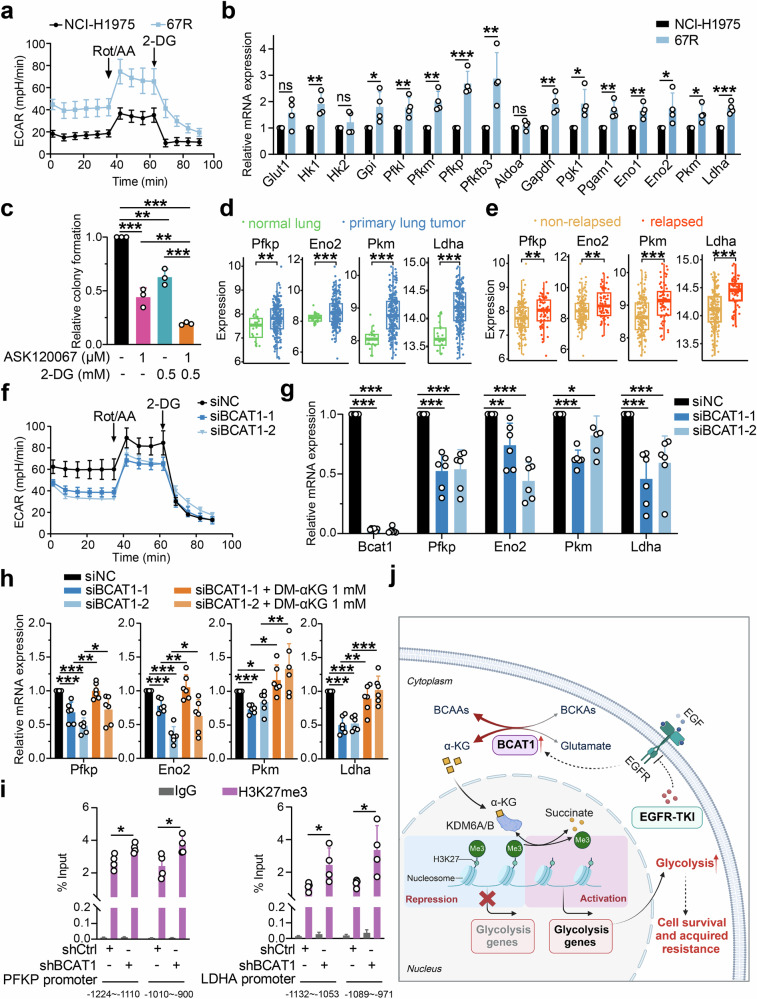
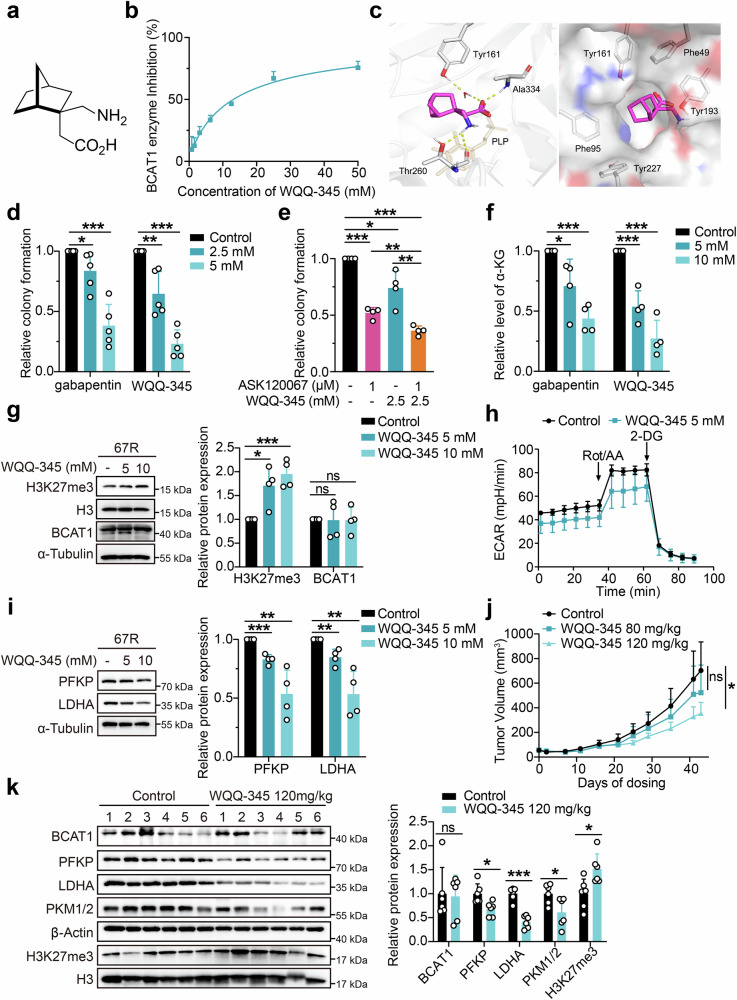
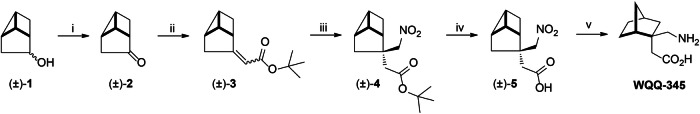
Third-generation EGFR tyrosine kinase inhibitors (TKIs), exemplified by osimertinib, have demonstrated promising clinical efficacy in the treatment of non-small cell lung cancer (NSCLC). Our previous work has identified ASK120067 as a novel third-generation EGFR TKI with remarkable antitumor effects that has undergone New Drug Application (NDA) submission in China. Despite substantial progress, acquired resistance to EGFR-TKIs remains a significant challenge, impeding the long-term effectiveness of therapeutic approaches. In this study, we conducted a comprehensive investigation utilizing high-throughput proteomics analysis on established TKI-resistant tumor models, and found a notable upregulation of branched-chain amino acid transaminase 1 (BCAT1) expression in both osimertinib- and ASK120067-resistant tumors compared with the parental TKI-sensitive NSCLC tumors. Genetic depletion or pharmacological inhibition of BCAT1 impaired the growth of resistant cells and partially re-sensitized tumor cells to EGFR TKIs. Mechanistically, upregulated BCAT1 in resistant cells reprogrammed branched-chain amino acid (BCAA) metabolism and promoted alpha ketoglutarate (α-KG)-dependent demethylation of lysine 27 on histone H3 (H3K27) and subsequent transcriptional derepression of glycolysis-related genes, thereby enhancing glycolysis and promoting tumor progression. Moreover, we identified WQQ-345 as a novel BCAT1 inhibitor exhibiting antitumor activity both in vitro and in vivo against TKI-resistant lung cancer with high BCAT1 expression. In summary, our study highlighted the crucial role of BCAT1 in mediating resistance to third-generation EGFR-TKIs through epigenetic activation of glycolysis in NSCLC, thereby supporting BCAT1 as a promising therapeutic target for the treatment of TKI-resistant NSCLC.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- 82273948/National Natural Science Foundation of China (National Science Foundation of China)
- 81903638/National Natural Science Foundation of China (National Science Foundation of China)
- SIMM2205KF-09/CAS | State Key Laboratory of Drug Research (State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
