

The genomic basis of childhood T-lineage acute lymphoblastic leukaemia
- PMID: 39143224
- PMCID: PMC11611067
- DOI: 10.1038/s41586-024-07807-0
The genomic basis of childhood T-lineage acute lymphoblastic leukaemia
Abstract
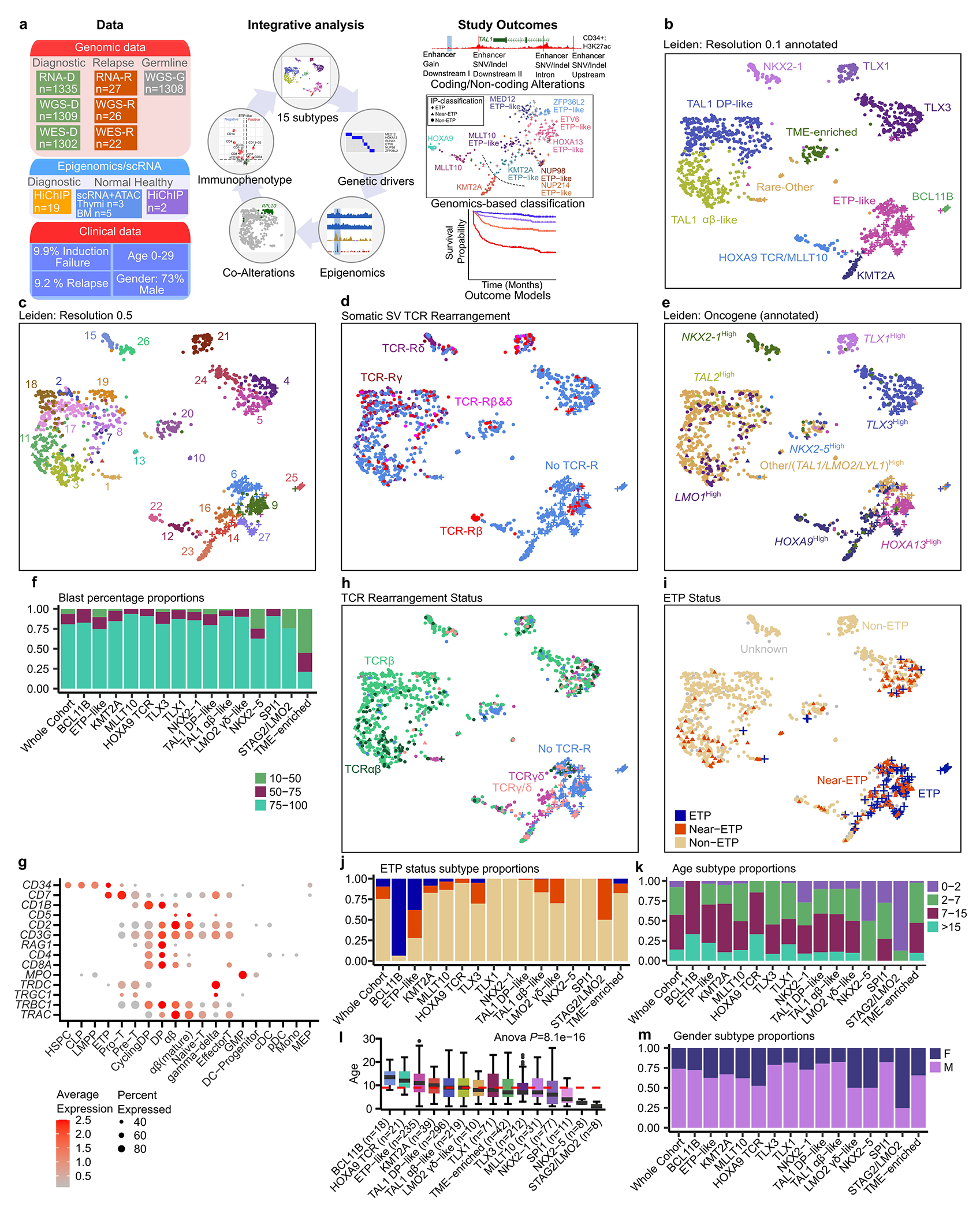
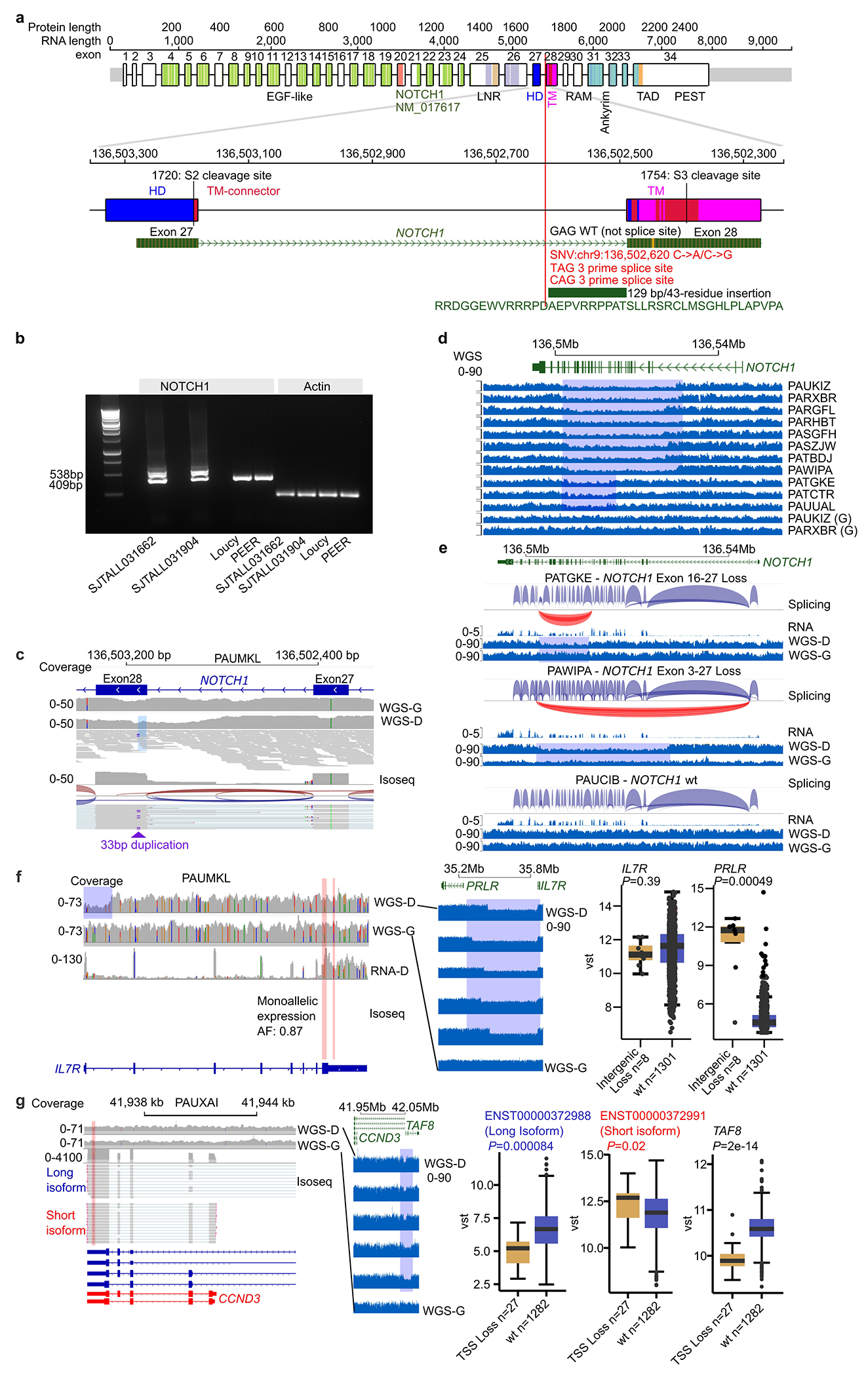
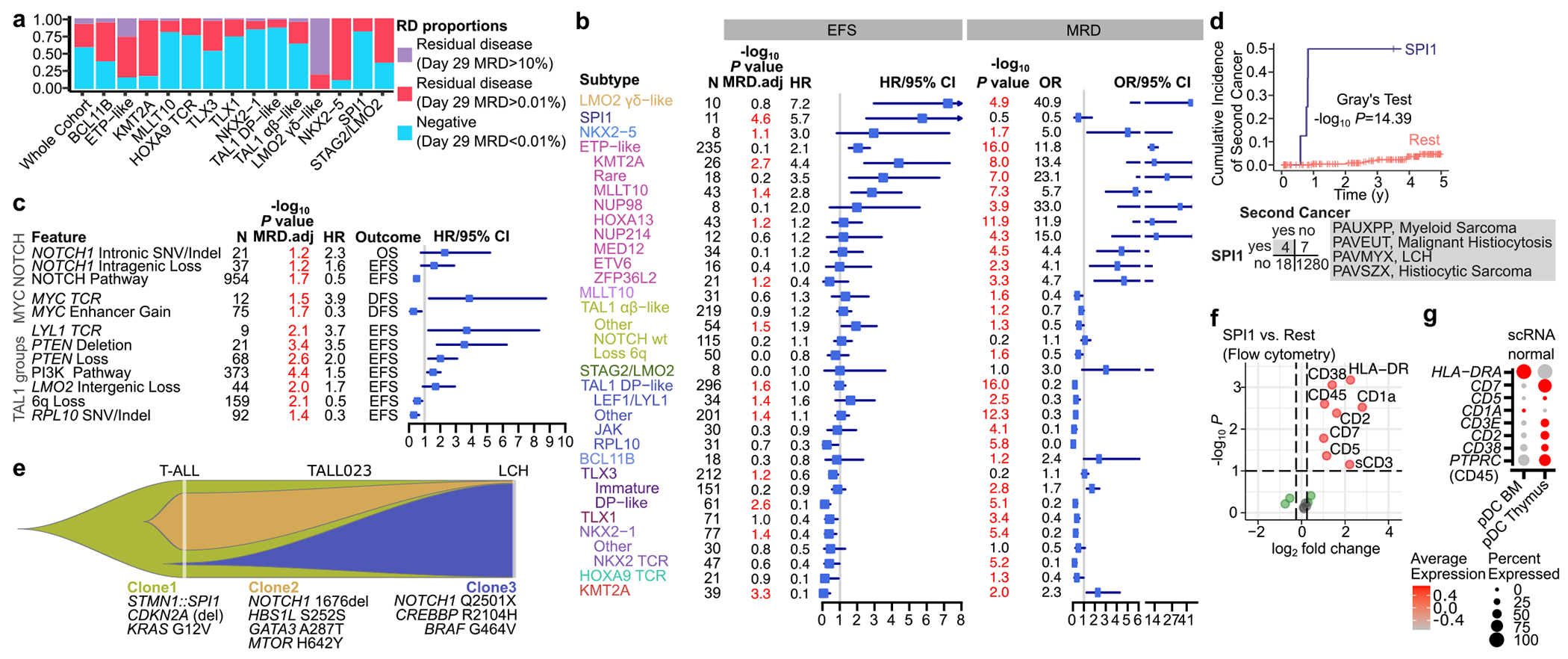
T-lineage acute lymphoblastic leukaemia (T-ALL) is a high-risk tumour1 that has eluded comprehensive genomic characterization, which is partly due to the high frequency of noncoding genomic alterations that result in oncogene deregulation2,3. Here we report an integrated analysis of genome and transcriptome sequencing of tumour and remission samples from more than 1,300 uniformly treated children with T-ALL, coupled with epigenomic and single-cell analyses of malignant and normal T cell precursors. This approach identified 15 subtypes with distinct genomic drivers, gene expression patterns, developmental states and outcomes. Analyses of chromatin topology revealed multiple mechanisms of enhancer deregulation that involve enhancers and genes in a subtype-specific manner, thereby demonstrating widespread involvement of the noncoding genome. We show that the immunophenotypically described, high-risk entity of early T cell precursor ALL is superseded by a broader category of 'early T cell precursor-like' leukaemia. This category has a variable immunophenotype and diverse genomic alterations of a core set of genes that encode regulators of hematopoietic stem cell development. Using multivariable outcome models, we show that genetic subtypes, driver and concomitant genetic alterations independently predict treatment failure and survival. These findings provide a roadmap for the classification, risk stratification and mechanistic understanding of this disease.
© 2024. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
D.T.T. received research funding from BEAM Therapeutics, NeoImmune Tech and serves on advisory boards for BEAM Therapeutics, Janssen, Servier, Sobi, and Jazz. D.T.T. has multiple patents pending on CAR-T. C.G.M. serves on the scientific advisory board and honoraria for Illumina, and received research funding from Pfizer, equity from Amgen and royalties from Cyrus. E.R Institutional research funding from Pfizer and serving on a Data and Safety Monitoring Board for Bristol Myers Squibb. I.I. Travel and accommodation expenses reimbursed by Mission Bio. I.I and P.P Consultancy fee by Arima Genomics. K.M.B research funding from Syndax.
Figures
















References
MeSH terms
Substances
Grants and funding
- U24 CA196173/CA/NCI NIH HHS/United States
- T32 CA236748/CA/NCI NIH HHS/United States
- K12 CA076931/CA/NCI NIH HHS/United States
- F32 CA254140/CA/NCI NIH HHS/United States
- T32 CA009615/CA/NCI NIH HHS/United States
- U10 CA180899/CA/NCI NIH HHS/United States
- U10 CA180886/CA/NCI NIH HHS/United States
- R35 CA197695/CA/NCI NIH HHS/United States
- R01 GM115634/GM/NIGMS NIH HHS/United States
- U54 CA243124/CA/NCI NIH HHS/United States
- K99 CA279756/CA/NCI NIH HHS/United States
- R01 CA193776/CA/NCI NIH HHS/United States
- R01 CA264837/CA/NCI NIH HHS/United States
- R03 CA256550/CA/NCI NIH HHS/United States
- U24 CA114766/CA/NCI NIH HHS/United States
- P30 CA021765/CA/NCI NIH HHS/United States
- K08 CA286762/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
