

Modulation of NOX2 causes obesity-mediated atrial fibrillation
- PMID: 39146015
- PMCID: PMC11405042
- DOI: 10.1172/JCI175447
Modulation of NOX2 causes obesity-mediated atrial fibrillation
Abstract
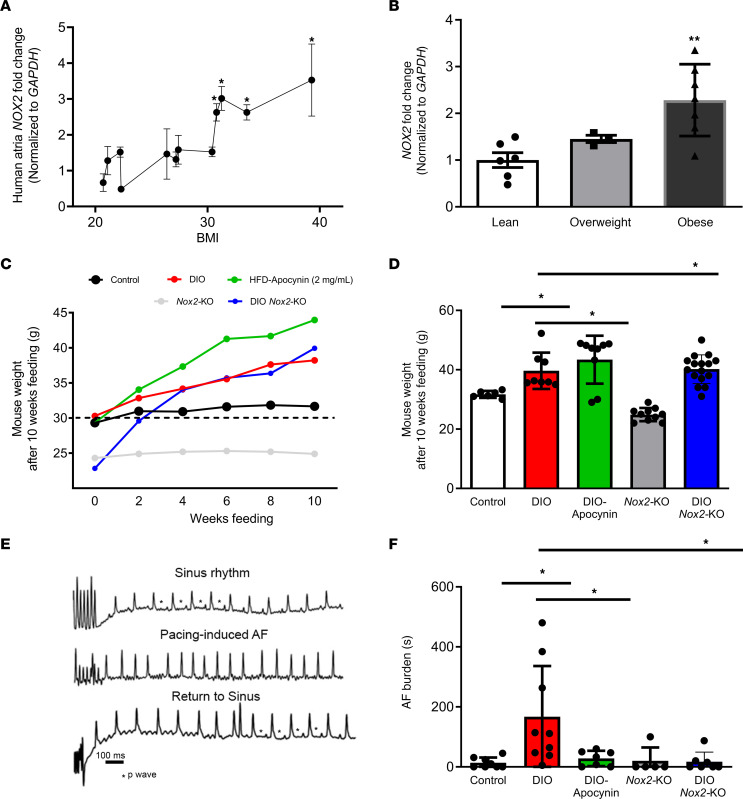
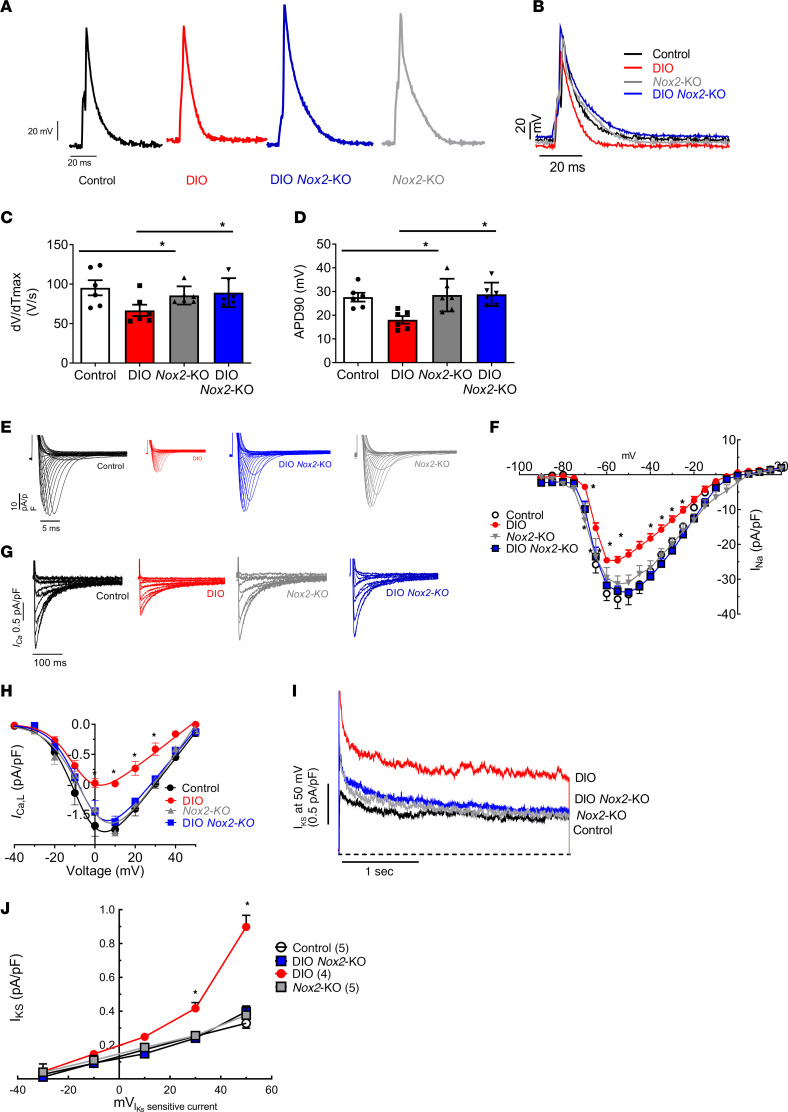
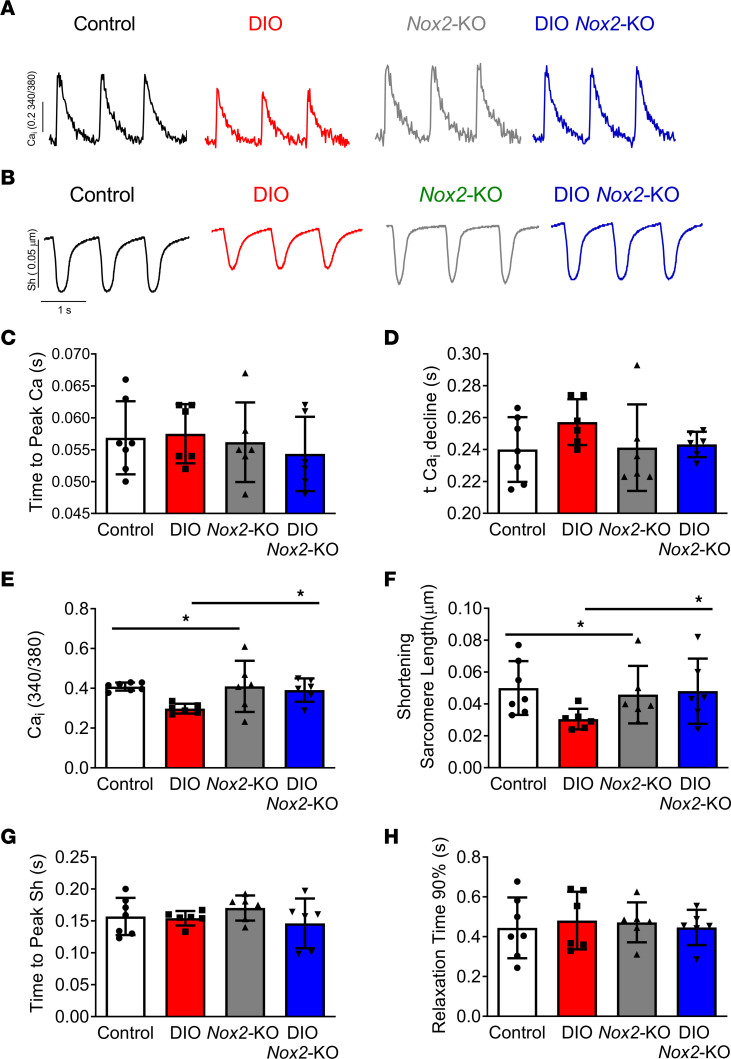
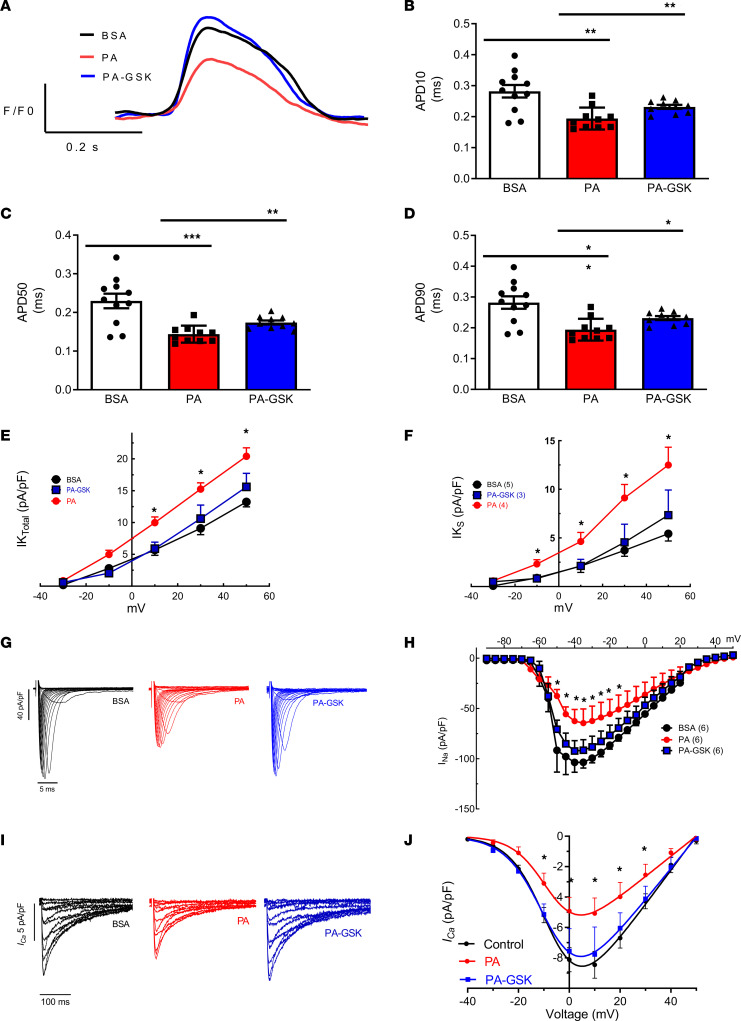
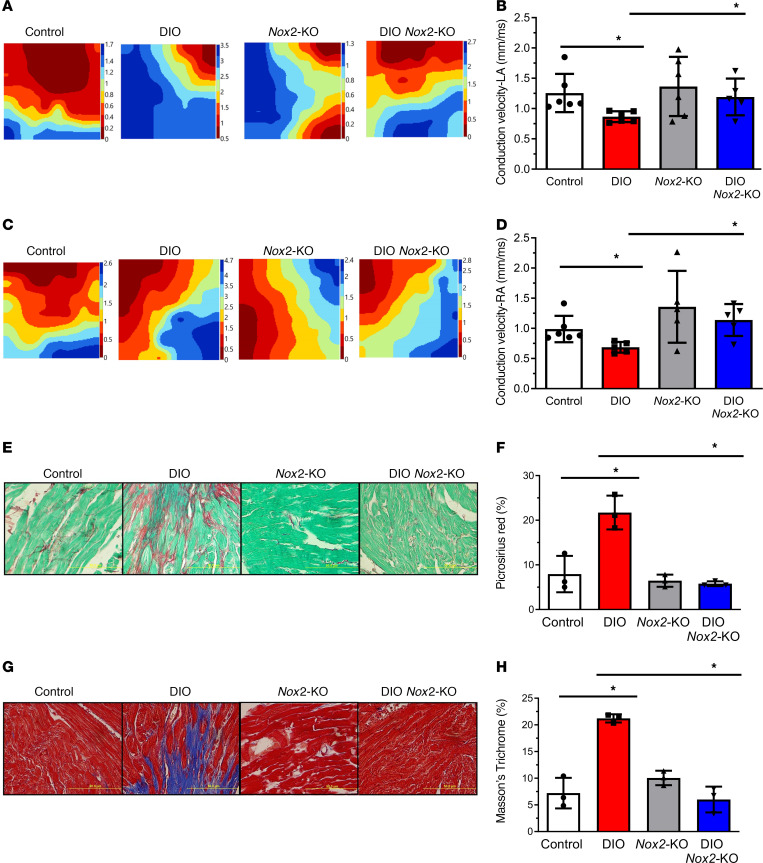
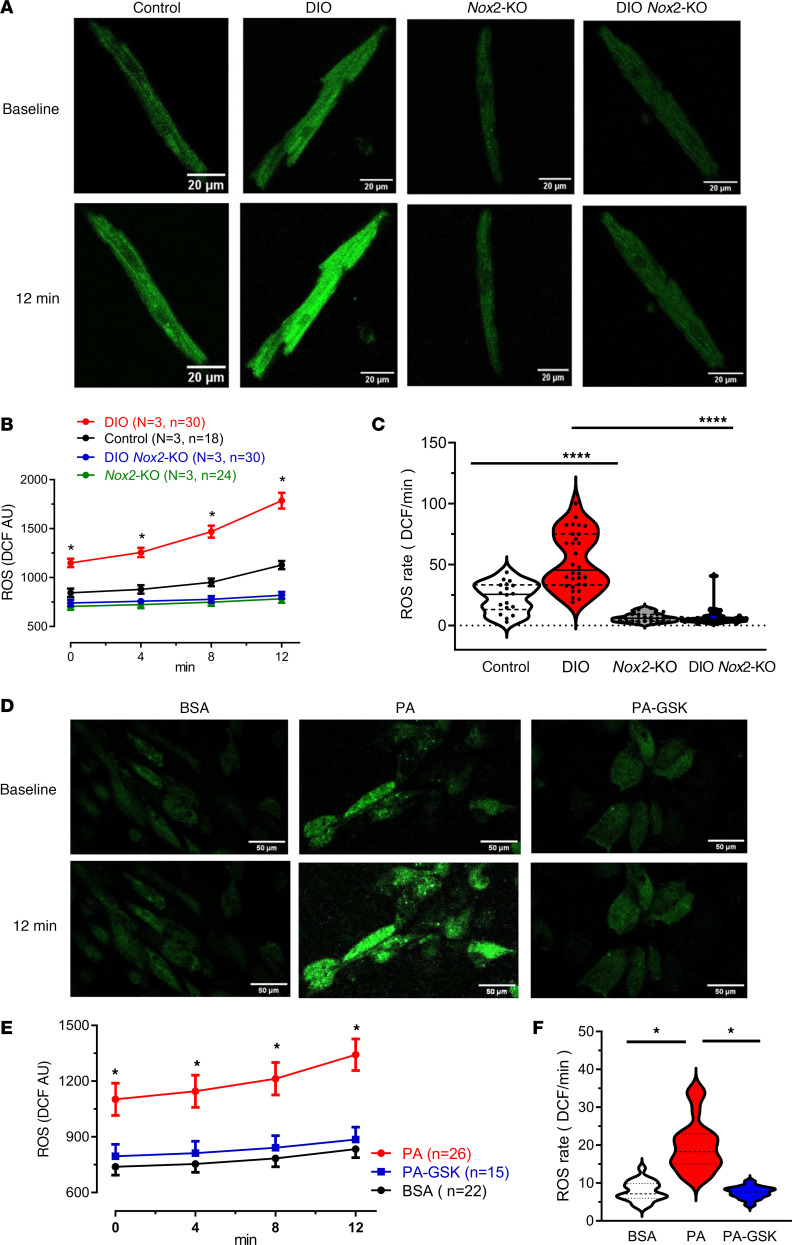
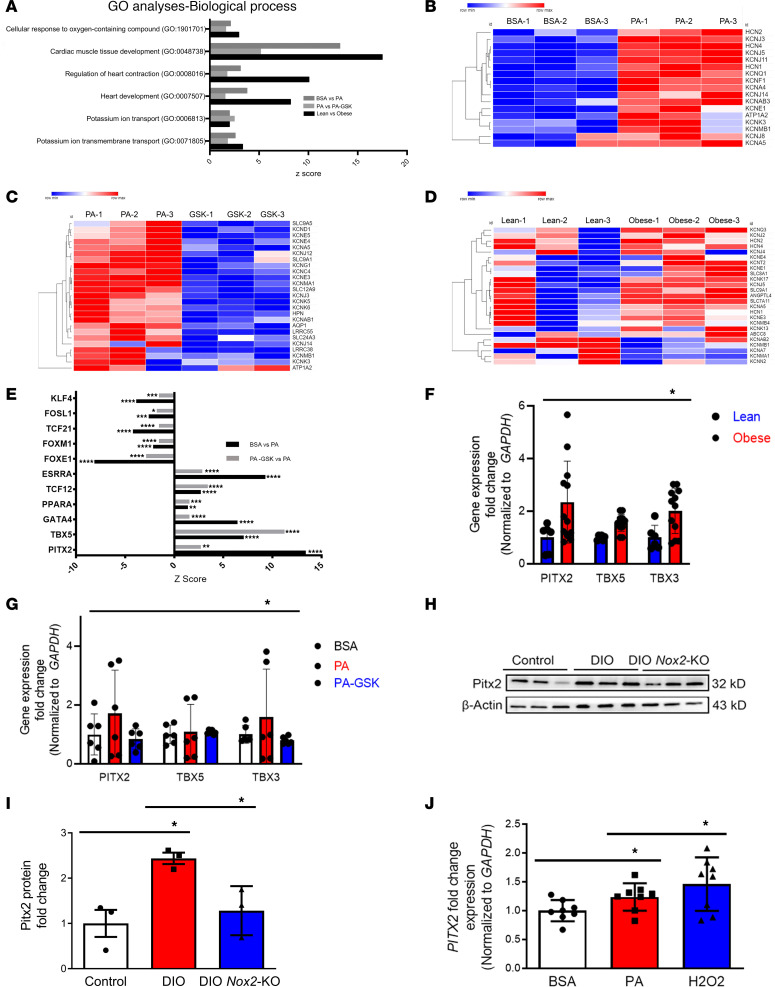
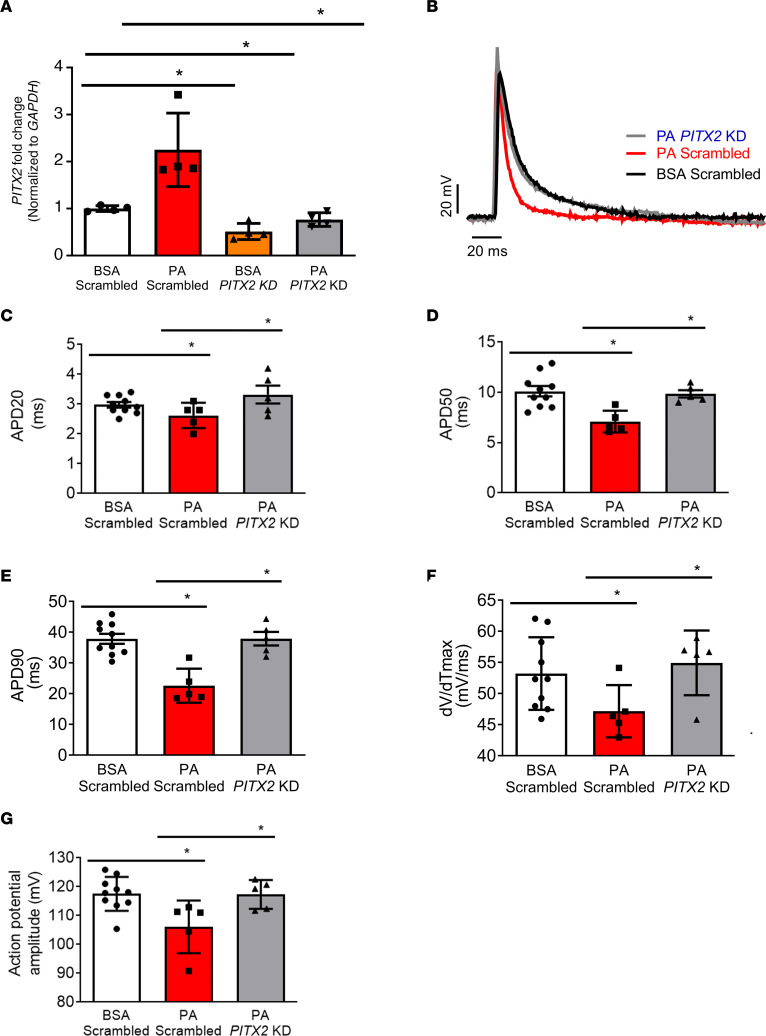
Obesity is linked to an increased risk of atrial fibrillation (AF) via increased oxidative stress. While NADPH oxidase 2 (NOX2), a major source of oxidative stress and reactive oxygen species (ROS) in the heart, predisposes to AF, the underlying mechanisms remain unclear. Here, we studied NOX2-mediated ROS production in obesity-mediated AF using Nox2-knockout mice and mature human induced pluripotent stem cell-derived atrial cardiomyocytes (hiPSC-aCMs). Diet-induced obesity (DIO) mice and hiPSC-aCMs treated with palmitic acid (PA) were infused with a NOX blocker (apocynin) and a NOX2-specific inhibitor, respectively. We showed that NOX2 inhibition normalized atrial action potential duration and abrogated obesity-mediated ion channel remodeling with reduced AF burden. Unbiased transcriptomics analysis revealed that NOX2 mediates atrial remodeling in obesity-mediated AF in DIO mice, PA-treated hiPSC-aCMs, and human atrial tissue from obese individuals by upregulation of paired-like homeodomain transcription factor 2 (PITX2). Furthermore, hiPSC-aCMs treated with hydrogen peroxide, a NOX2 surrogate, displayed increased PITX2 expression, establishing a mechanistic link between increased NOX2-mediated ROS production and modulation of PITX2. Our findings offer insights into possible mechanisms through which obesity triggers AF and support NOX2 inhibition as a potential novel prophylactic or adjunctive therapy for patients with obesity-mediated AF.
Keywords: Arrhythmias; Cardiology.
Conflict of interest statement
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
