

Cocaine-derived hippuric acid activates mtDNA-STING signaling in alcoholic liver disease: Implications for alcohol and cocaine co-abuse
- PMID: 39147926
- PMCID: PMC11327214
- DOI: 10.1007/s10565-024-09901-5
Cocaine-derived hippuric acid activates mtDNA-STING signaling in alcoholic liver disease: Implications for alcohol and cocaine co-abuse
Abstract
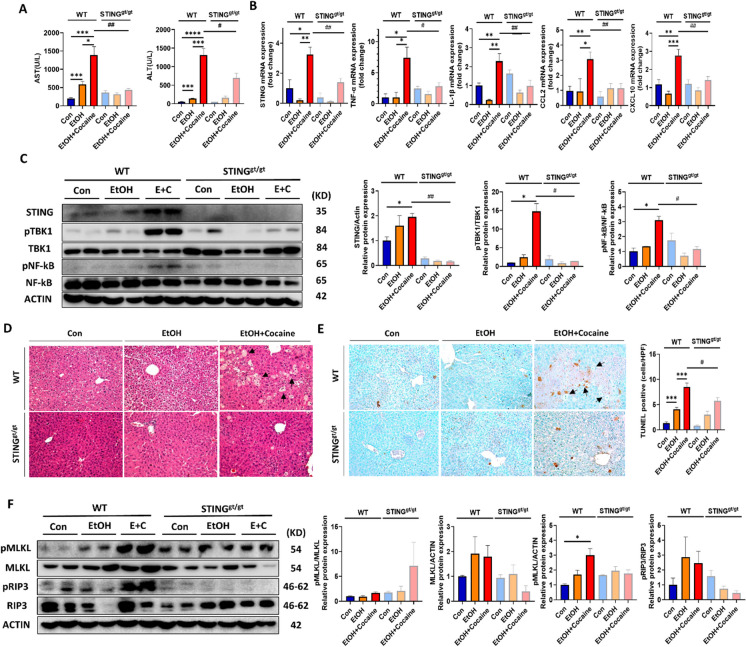
The simultaneous abuse of alcohol-cocaine is known to cause stronger and more unpredictable cellular damage in the liver, heart, and brain. However, the mechanistic crosstalk between cocaine and alcohol in liver injury remains unclear. The findings revealed cocaine-induced liver injury and inflammation in both marmosets and mice. Of note, co-administration of cocaine and ethanol in mice causes more severe liver damage than individual treatment. The metabolomic analysis confirmed that hippuric acid (HA) is the most abundant metabolite in marmoset serum after cocaine consumption and that is formed in primary marmoset hepatocytes. HA, a metabolite of cocaine, increases mitochondrial DNA leakage and subsequently increases the production of proinflammatory factors via STING signaling in Kupffer cells (KCs). In addition, conditioned media of cocaine-treated KC induced hepatocellular necrosis via alcohol-induced TNFR1. Finally, disruption of STING signaling in vivo ameliorated co-administration of alcohol- and cocaine-induced liver damage and inflammation. These findings postulate intervention of HA-STING-TNFR1 axis as a novel strategy for treatment of alcohol- and cocaine-induced excessive liver damage.
Keywords: Cocaine; Hippuric acid; Liver disease; Mitochondrial DNA leakage; STING; TNFR1.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures







References
-
- Amoedo ML, Craver L, Marco MP, Fernandez E. Cocaine-induced acute renal failure without rhabdomyolysis. Nephrol Dial Transplant. 1999;14:2970–1. 10.1093/ndt/14.12.2970. - PubMed
-
- An L, Wang X, Cederbaum AI. Cytokines in alcoholic liver disease. Arch Toxicol. 2012;86:1337–48. 10.1007/s00204-012-0814-6. - PubMed
-
- Bailey DN. Plasma cocaethylene concentrations in patients treated in the emergency room or trauma unit. Am J Clin Pathol. 1993;99:123–7. 10.1093/ajcp/99.2.123. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 2017R1A5A2015541/National Research Foundation of Korea
- 2017R1A5A2015541/National Research Foundation of Korea
- 2017R1A5A2015541/National Research Foundation of Korea
- 2017R1A5A2015541/National Research Foundation of Korea
- 2017R1A5A2015541/National Research Foundation of Korea
- 2017R1A5A2015541/National Research Foundation of Korea
- 2017R1A5A2015541/National Research Foundation of Korea
- 2017R1A5A2015541/National Research Foundation of Korea
- 2017R1A5A2015541/National Research Foundation of Korea
- 2017R1A5A2015541/National Research Foundation of Korea
- 2017R1A5A2015541/National Research Foundation of Korea
- 2017R1A5A2015541/National Research Foundation of Korea
- 2021RIS-001/Regional Innovation Strategy (RIS) of National Research Foundation of Korea
- 2021RIS-001/Regional Innovation Strategy (RIS) of National Research Foundation of Korea
- 2021RIS-001/Regional Innovation Strategy (RIS) of National Research Foundation of Korea
- 2021RIS-001/Regional Innovation Strategy (RIS) of National Research Foundation of Korea
- 2021RIS-001/Regional Innovation Strategy (RIS) of National Research Foundation of Korea
- 2021RIS-001/Regional Innovation Strategy (RIS) of National Research Foundation of Korea
- 2021RIS-001/Regional Innovation Strategy (RIS) of National Research Foundation of Korea
- 2021RIS-001/Regional Innovation Strategy (RIS) of National Research Foundation of Korea
- 2021RIS-001/Regional Innovation Strategy (RIS) of National Research Foundation of Korea
- 2021RIS-001/Regional Innovation Strategy (RIS) of National Research Foundation of Korea
- 2021RIS-001/Regional Innovation Strategy (RIS) of National Research Foundation of Korea
- 2021RIS-001/Regional Innovation Strategy (RIS) of National Research Foundation of Korea
- 20182MFDS425/Korean Food and Drug Administration
- 20182MFDS425/Korean Food and Drug Administration
- 20182MFDS425/Korean Food and Drug Administration
- 20182MFDS425/Korean Food and Drug Administration
- 20182MFDS425/Korean Food and Drug Administration
- 20182MFDS425/Korean Food and Drug Administration
- 20182MFDS425/Korean Food and Drug Administration
- 20182MFDS425/Korean Food and Drug Administration
- 20182MFDS425/Korean Food and Drug Administration
- 20182MFDS425/Korean Food and Drug Administration
- 20182MFDS425/Korean Food and Drug Administration
- 20182MFDS425/Korean Food and Drug Administration
LinkOut - more resources
Full Text Sources
Research Materials
