

Exploring the influence of H-bonding and ligand constraints on thiolate ligated non-heme iron mediated dioxygen activation
- PMID: 39148773
- PMCID: PMC11325341
- DOI: 10.1039/d4sc02787f
Exploring the influence of H-bonding and ligand constraints on thiolate ligated non-heme iron mediated dioxygen activation
Abstract
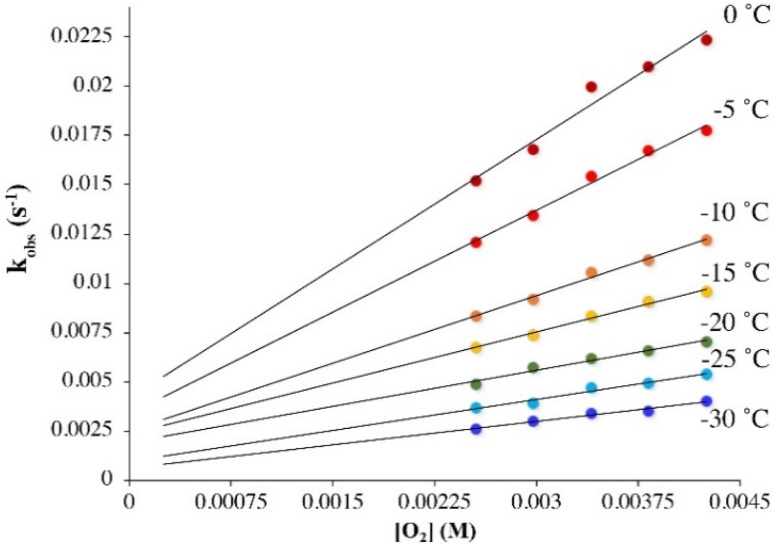
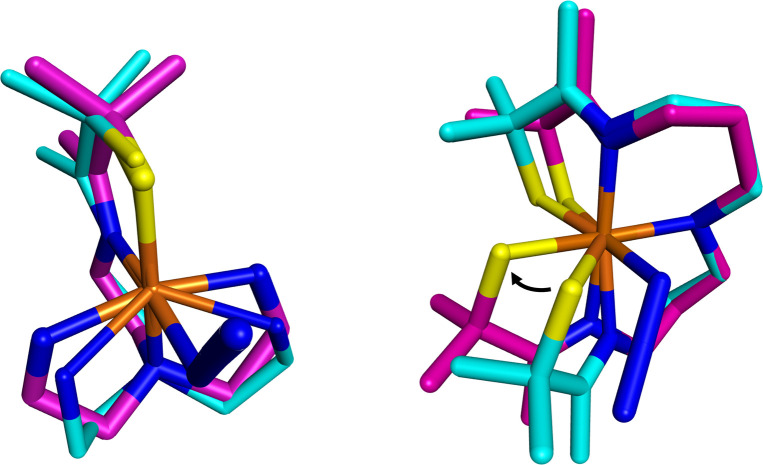
Converting triplet dioxygen into a powerful oxidant is fundamentally important to life. The study reported herein quantitatively examines the formation of a well-characterized, reactive, O2-derived thiolate ligated FeIII-superoxo using low-temperature stopped-flow kinetics. Comparison of the kinetic barriers to the formation of this species via two routes, involving either the addition of (a) O2 to [FeII(S2 Me2N3(Pr,Pr))] (1) or (b) superoxide to [FeIII(S2 Me2N3(Pr,Pr))]+ (3) is shown to provide insight into the mechanism of O2 activation. Route (b) was shown to be significantly slower, and the kinetic barrier 14.9 kJ mol-1 higher than route (a), implying that dioxygen activation involves inner-sphere, as opposed to outer sphere, electron transfer from Fe(ii). H-bond donors and ligand constraints are shown to dramatically influence O2 binding kinetics and reversibility. Dioxygen binds irreversibly to [FeII(S2 Me2N3(Pr,Pr))] (1) in tetrahydrofuran, but reversibly in methanol. Hydrogen bonding decreases the ability of the thiolate sulfur to stabilize the transition state and the FeIII-superoxo, as shown by the 10 kJ mol-1 increase in the kinetic barrier to O2 binding in methanol vs. tetrahydrofuran. Dioxygen release from [FeIII(S2 Me2N3(Pr,Pr))O2] (2) is shown to be 24 kJ mol-1 higher relative to previously reported [FeIII(SMe2N4(tren))(O2)]+ (5), the latter of which contains a more flexible ligand. These kinetic results afford an experimentally determined reaction coordinate that illustrates the influence of H-bonding and ligand constraints on the kinetic barrier to dioxygen activation an essential step in biosynthetic pathways critical to life.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures











References
-
- Jeong D. Selverstone Valentine J. Cho J. Bio-inspired mononuclear nonheme metal peroxo complexes: synthesis, structures and mechanistic studies toward understanding enzymatic reactions. Coordin. Chem. Rev. 2023;480:215021. doi: 10.1016/j.ccr.2023.215021. - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials