

DSS1 restrains BRCA2's engagement with dsDNA for homologous recombination, replication fork protection, and R-loop homeostasis
- PMID: 39152168
- PMCID: PMC11329725
- DOI: 10.1038/s41467-024-51557-6
DSS1 restrains BRCA2's engagement with dsDNA for homologous recombination, replication fork protection, and R-loop homeostasis
Abstract
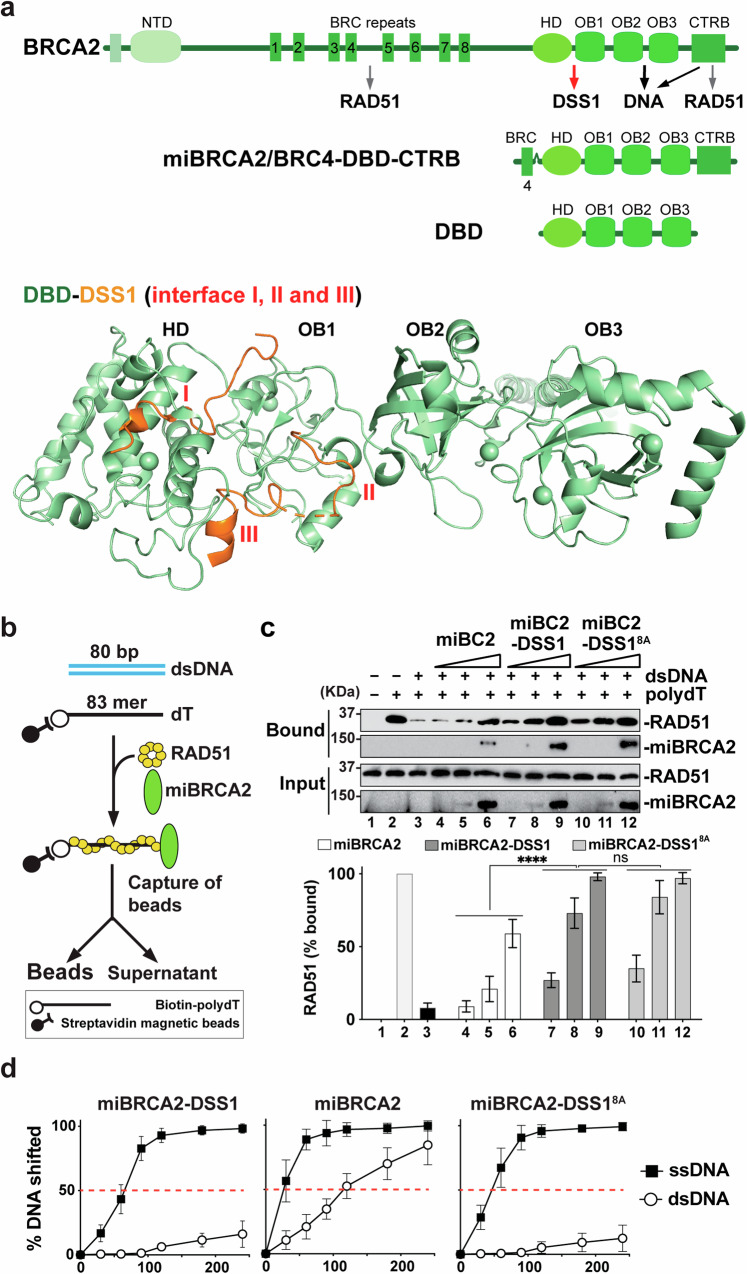
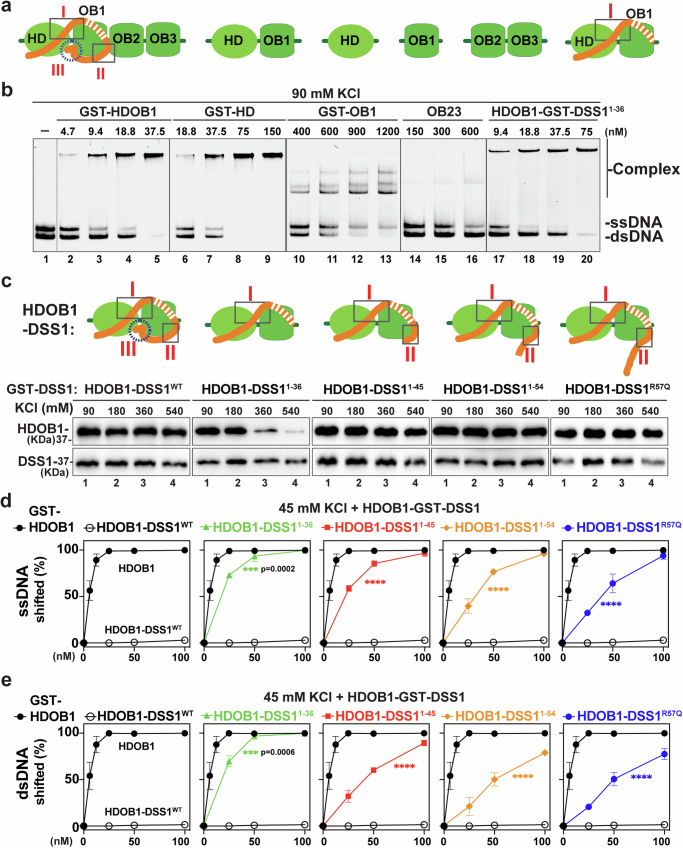
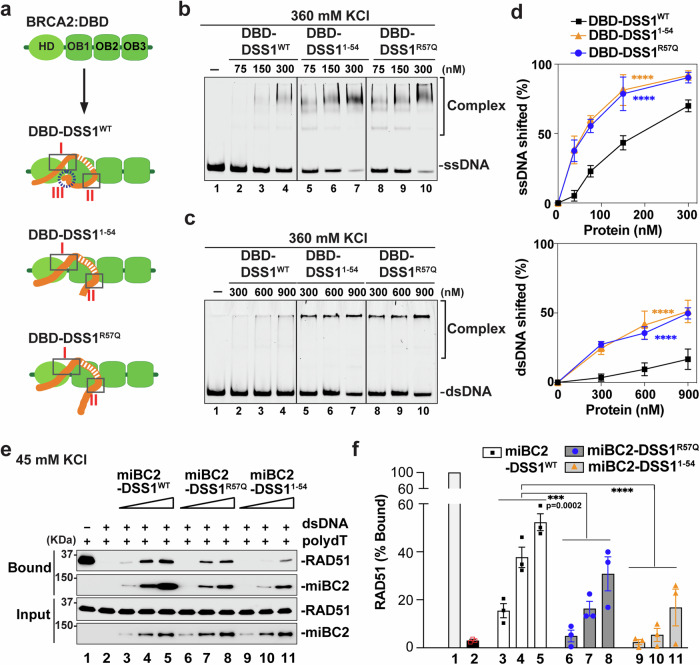
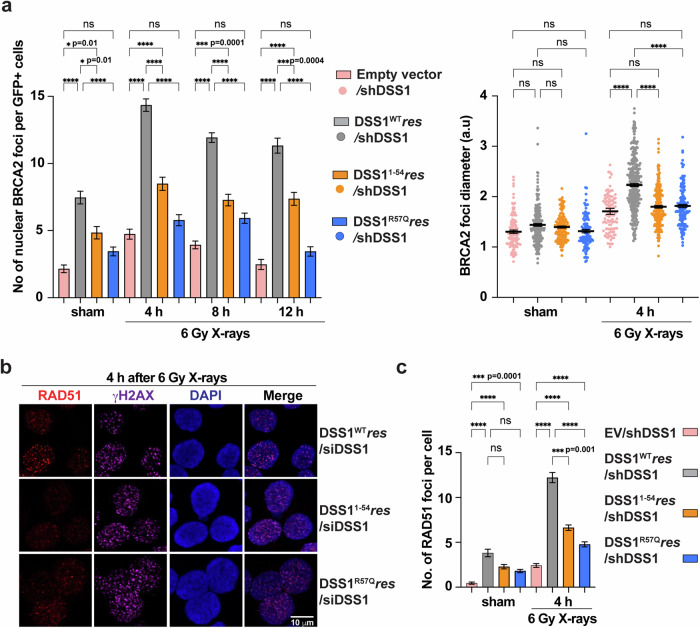
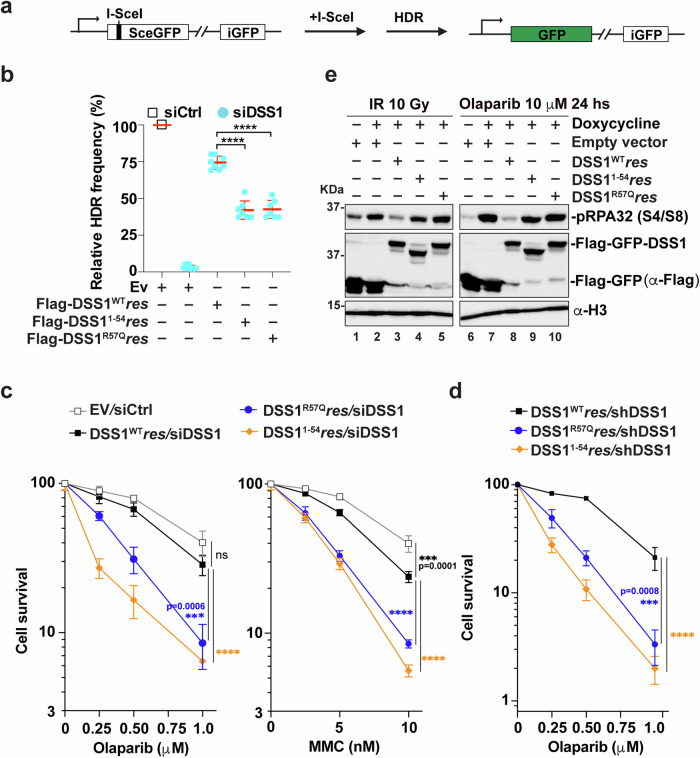
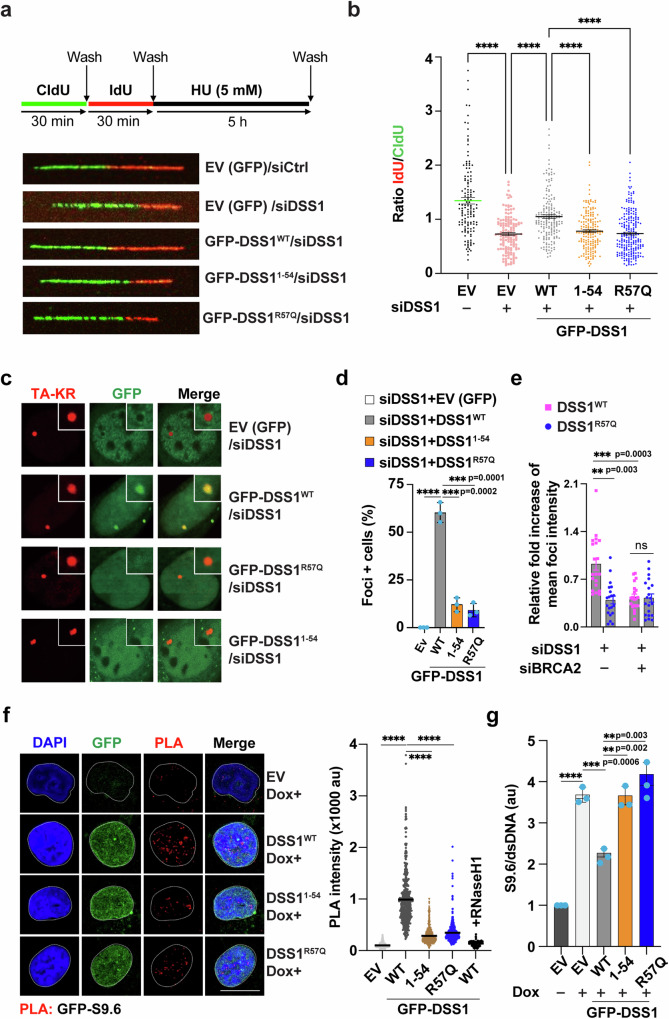
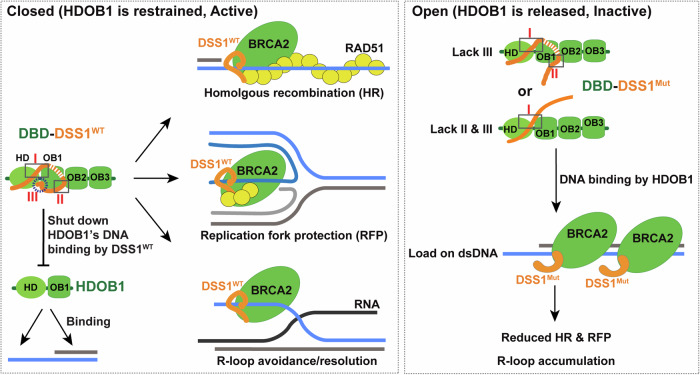
DSS1, essential for BRCA2-RAD51 dependent homologous recombination (HR), associates with the helical domain (HD) and OB fold 1 (OB1) of the BRCA2 DSS1/DNA-binding domain (DBD) which is frequently targeted by cancer-associated pathogenic variants. Herein, we reveal robust ss/dsDNA binding abilities in HD-OB1 subdomains and find that DSS1 shuts down HD-OB1's DNA binding to enable ssDNA targeting of the BRCA2-RAD51 complex. We show that C-terminal helix mutations of DSS1, including the cancer-associated R57Q mutation, disrupt this DSS1 regulation and permit dsDNA binding of HD-OB1/BRCA2-DBD. Importantly, these DSS1 mutations impair BRCA2/RAD51 ssDNA loading and focus formation and cause decreased HR efficiency, destabilization of stalled forks and R-loop accumulation, and hypersensitize cells to DNA-damaging agents. We propose that DSS1 restrains the intrinsic dsDNA binding of BRCA2-DBD to ensure BRCA2/RAD51 targeting to ssDNA, thereby promoting optimal execution of HR, and potentially replication fork protection and R-loop suppression.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
- R01 CA246807/CA/NCI NIH HHS/United States
- R01GM141091/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- RP210102/Cancer Prevention and Research Institute of Texas (Cancer Prevention Research Institute of Texas)
- R01 CA262227/CA/NCI NIH HHS/United States
- R01 GM141091/GM/NIGMS NIH HHS/United States
- P01 CA250957/CA/NCI NIH HHS/United States
- R01 CA282939/CA/NCI NIH HHS/United States
- R01 GM144559/GM/NIGMS NIH HHS/United States
- R01CA138804/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- a Young Investigator Award/Max and Minnie Tomerlin Voelcker Fund (Voelcker Fund)
- V scholar Award/V Foundation for Cancer Research (V Foundation)
- R01 GM135651/GM/NIGMS NIH HHS/United States
- R01CA246807/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01CA262227/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01CA268641/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01 CA138804/CA/NCI NIH HHS/United States
- R01 CA268641/CA/NCI NIH HHS/United States
- R01CA282939/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- RSG-22-721675-01-DMC/American Cancer Society (American Cancer Society, Inc.)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
