

Case report: Comprehensive exploration of a novel PFKM mutation in glycogen storage disease Type VII
- PMID: 39156960
- PMCID: PMC11327043
- DOI: 10.3389/fgene.2024.1422908
Case report: Comprehensive exploration of a novel PFKM mutation in glycogen storage disease Type VII
Abstract
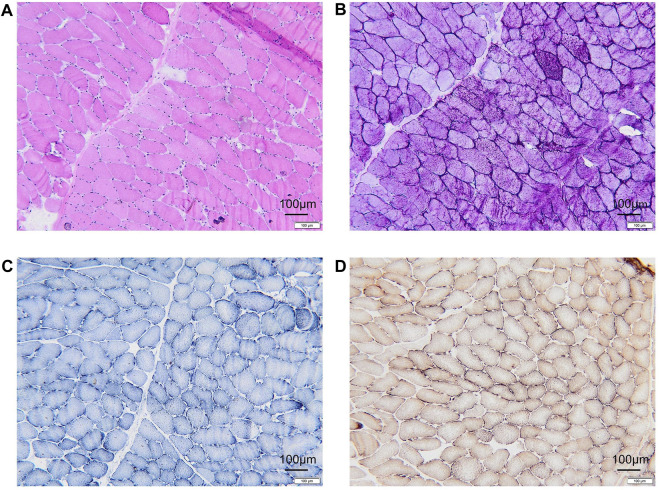
Glycogen Storage Disease Type VII (GSD VII) is a rare glycogen metabolism disorder resulting from mutations in the PFKM gene, inherited in an autosomal recessive manner. It is characterized by exercise intolerance, muscle cramps, myoglobinuria, compensatory hemolysis, and later onset de novo myasthenia and mild myopathy, contributing to its clinical heterogeneity and diagnostic challenges. Here, we report a rare case of a 17-year-old Chinese woman exhibiting substantial muscle weakness and compensated hemolysis. Muscle biopsies showed glycogen deposition, and blood tests showed hyperuricemia and significantly elevated creatine kinase. Whole genome sequencing (WGS) and whole exome sequencing (WES) identified two compound heterozygous mutations in the PFKM (NM_000289.6) gene: c.626G>A and c.1376G>A in exons 7 and 15, respectively. According to the clinical presentation, diagnostic examination, and WES results, the patient was finally diagnosed with GSDVII. The discovery of these two new PFKM mutations expands the genetic spectrum, and understanding the clinical manifestations of these mutations is critical to preventing diagnostic delays and timely intervention and treatment.
Keywords: PFKM; case report; compound heterozygous mutations; glycogen storage disease Type VII; hemolysis.
Copyright © 2024 Chen, Wang, Ji, Fang, Chang and Liu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
Publication types
LinkOut - more resources
Full Text Sources