

Comprehensive molecular profiling of multiple myeloma identifies refined copy number and expression subtypes
- PMID: 39160255
- PMCID: PMC11387199
- DOI: 10.1038/s41588-024-01853-0
Comprehensive molecular profiling of multiple myeloma identifies refined copy number and expression subtypes
Erratum in
-
Author Correction: Comprehensive molecular profiling of multiple myeloma identifies refined copy number and expression subtypes.Nat Genet. 2025 Jul;57(7):1789. doi: 10.1038/s41588-025-02251-w. Nat Genet. 2025. PMID: 40490515 Free PMC article. No abstract available.
Abstract
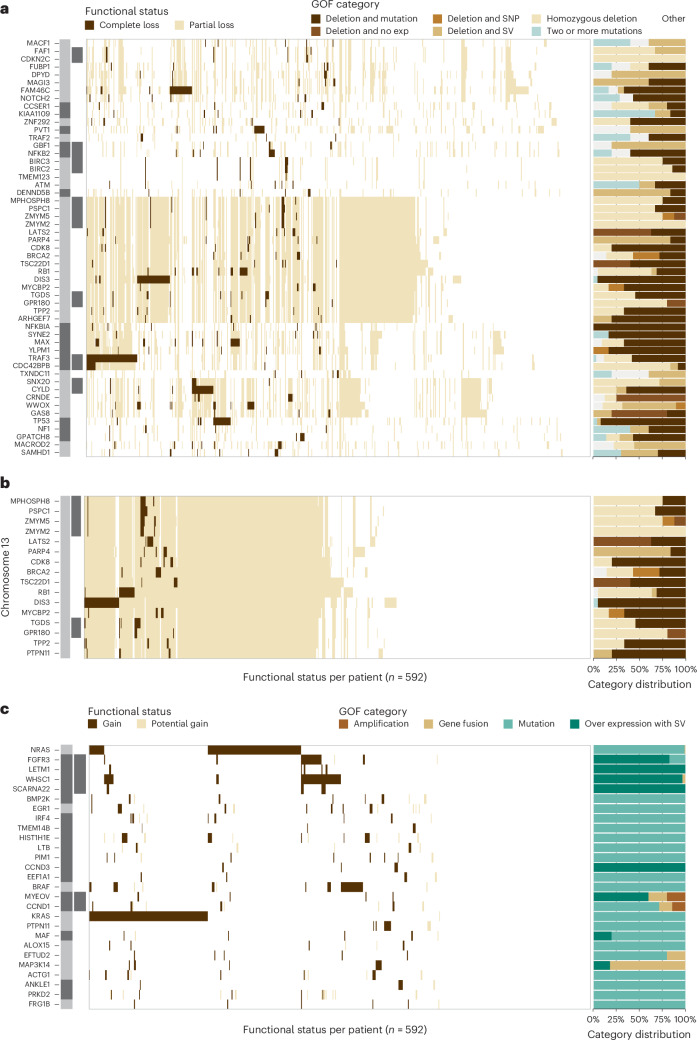
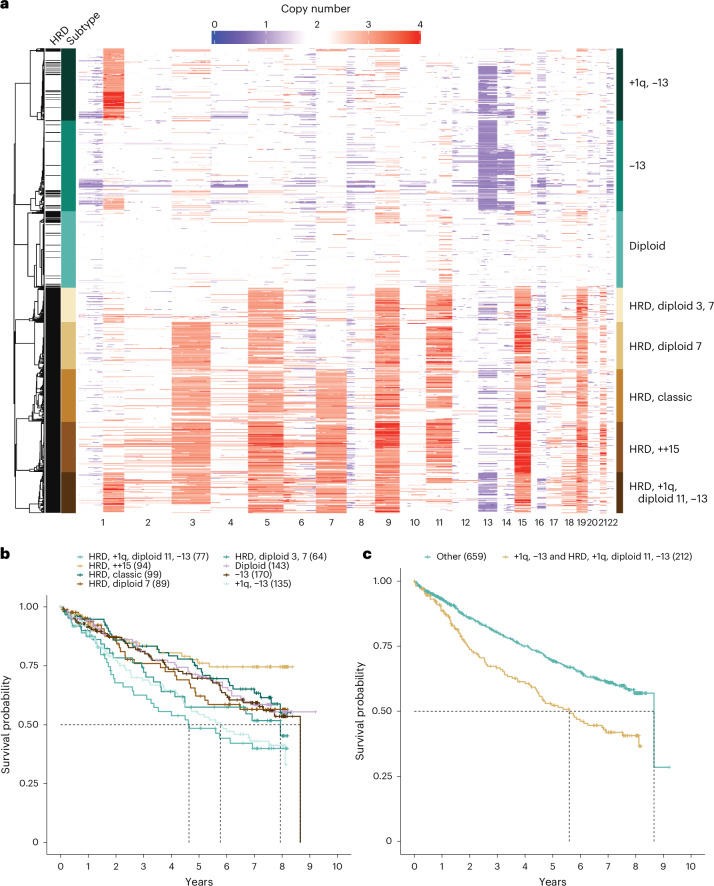
Multiple myeloma is a treatable, but currently incurable, hematological malignancy of plasma cells characterized by diverse and complex tumor genetics for which precision medicine approaches to treatment are lacking. The Multiple Myeloma Research Foundation's Relating Clinical Outcomes in Multiple Myeloma to Personal Assessment of Genetic Profile study ( NCT01454297 ) is a longitudinal, observational clinical study of newly diagnosed patients with multiple myeloma (n = 1,143) where tumor samples are characterized using whole-genome sequencing, whole-exome sequencing and RNA sequencing at diagnosis and progression, and clinical data are collected every 3 months. Analyses of the baseline cohort identified genes that are the target of recurrent gain-of-function and loss-of-function events. Consensus clustering identified 8 and 12 unique copy number and expression subtypes of myeloma, respectively, identifying high-risk genetic subtypes and elucidating many of the molecular underpinnings of these unique biological groups. Analysis of serial samples showed that 25.5% of patients transition to a high-risk expression subtype at progression. We observed robust expression of immunotherapy targets in this subtype, suggesting a potential therapeutic option.
© 2024. The Author(s).
Conflict of interest statement
S.S. is currently employed by Janssen. A.C. has received research funding from Amgen, BMS, Janssen, Seattle Genetics and Takeda, and is a speaker/consultant/advisory board member for Abbvie, Amgen, Antengene, BMS, Genentech, GSK, Janssen, Karyopharm, Sanofi, Seattle Genetics, Secura Bio, Shattuck Labs and Takeda. S.J. is a speaker/consultant/advisory board member for BMS, Caribou Biosciences, DMC, Janssen, Regeneron, Sanofi and Takeda. D.S.S. has received research funding from Amgen, BMS, Janssen, Karyopharm, Novartis and Takeda, and is a speaker/consultant/advisory board member for Amgen, BMS, Celularity Scientific, Janssen, Karyopharm, Novartis and Takeda. R.V. has received research funding from BMS, Sanofi and Takeda, and is a speaker/consultant/advisory board member for BMS, Harpoon, Janssen Karyopharm, Legend Biotech, Pfizer, Sanofi and Takeda. A.J. is a speaker/consultant/advisory board member for Abbvie, BMS, Amgen, GSK, Janssen and Sanofi. R.N. has received research funding from BMS, GSK, Janssen, Karyopharm, Pfizer, Regeneron and Takeda, and is a speaker/consultant/advisory board member for BMS, GSK, Janssen, Karyopharm, Pfizer, Regeneron, Sanofi and Takeda. D.L. has received research funding from Annexon Biosciences, Alpine Immune, Abbvie, Astex Pharmaceuticals, Baxalta, BeiGene, Bioverativ, CSL Behring, BMS, Delta-Fly Pharma, Exact Sciences, Forma Therapeutics, Global Blood Therapeutics, Immunovant, Incycte, Janssen, NeoImmuneTech, Novartis, Novo Nordisk, Partner Therapeutics, Pharm-Olam, Principia Biopharma, Salix Pharmaceuticals, Sanofi, Takeda and Vifor Pharma. J.B. has received research funding from AbbVie, Acetylon, Amgen, BMS, C4 Therapeutics, CARSgen, Cartesian, Celularity, CRISPR Therapeutics, EMD Serono, Fate Therapeutics, Genentech, GSK, Ichnos Sciences, Incyte, Janssen, Karyopharm, Lilly, Novartis, Poseida, Sanofi, Takeda, Teva and 2seventy bio, and is a speaker/consultant/advisory board member for BMS, CRISPR Therapeutics, Janssen, Kite Pharma, Legend Biotech, Roche and Takeda. M.L. is a speaker/consultant/advisory board member for Abbvie, Amgen, AstraZeneca, BeiGene, BMS, Gilead, Genmab, Janssen, Jazz, Karyopharm, Morphosys, Novartis, Seagen, Sellas, Sobi, Sanofi and Takeda. S.Z.U. has received research funding from AbbVie, Amgen, Array Biopharma, BMS, EdoPharma, Gilead Sciences, GSK, Janssen, K36 Therapeutics, Merck, Moderna, Pharmacyclics, Sanofi, Seattle Genetics, SkylineDX and Takeda, and is a speaker/consultant/advisory board member for BMS, Genentech, Gilead Sciences, GSK, Janssen, Novartis, Oncopeptides, Sanofi, Seattle Genetics, Secura Bio, SkylineDX, Takeda and TeneoBio. M.D’A. has received research funding from Janssen and is a speaker/consultant/advisory board member for Adaptive Biotechnology, BMS, GSK, Janssen and Sanofi. M.D. is currently employed by the funder (MMRF). J.R. is currently employed by the funder (MMRF). K.C.A. is a speaker/consultant/advisory board member for Amgen, AstraZeneca, Daewoong, Dynamic Cell Therapies, Janssen, Pfizer, Mana Therapeutics, Oncopep, Precision Biosciences, Starton and Window Therapeutics, and is an equity holder in C4 Therapeutics, Dynamic Cell Therapies, NextRNA, Oncopep, Raqia, Starton and Window Therapeutics. H.J.C. is currently employed by the funder (MMRF) and has received research funding from BMS and Takeda. D.A. was previously employed by the funder (MMRF) and is currently employed by AstraZeneca. S.L. has received research funding from BMS, Janssen, Novartis and Takeda, and is a speaker/consultant/advisory board member for AbbVie, Amgen, BMS, Genentech, GSK, Janssen, Novartis, Pfizer, TG Therapeutics and Takeda. J.J.K. has received research funding from Amgen, Genentech and Janssen, and is a speaker/consultant/advisory board member for Janssen. The other authors declare no competing interests.
Figures













References
-
- Venti, G., Mecucci, C., Donti, E. & Tabilio, A. Translocation t(11;14) and trisomy 11q13-qter in multiple myeloma. Ann. Genet.27, 53–55 (1984). - PubMed
