

Avalglucosidase alfa in infantile-onset Pompe disease: A snapshot of real-world experience in Italy
- PMID: 39161458
- PMCID: PMC11332206
- DOI: 10.1016/j.ymgmr.2024.101126
Avalglucosidase alfa in infantile-onset Pompe disease: A snapshot of real-world experience in Italy
Abstract
Introduction: Infantile-onset Pompe disease (IOPD) is due to mutations in the GAA gene leading to profound deficiency of the lysosomal enzyme α-1,4-glucosidase. The disease is characterized by severe hypotonia, hypertrophic cardiomyopathy, macroglossia, and liver enlargement with onset in the first months of life. In the late-onset form (LOPD), muscle signs predominate with a clinical picture resembling muscle dystrophies. Enzyme replacement therapy with alglucosidase alfa (rhGAA) has been available since 2006 and patients treated with the enzyme show improved outcomes. Nevertheless, there is evidence that some patients have a suboptimal response or, after an initial improvement, reach a plateau with stabilization of the clinical picture. Thus, a new enzyme formulation, avalglucosidase alfa (neoGAA), with a higher degree of mannosylation, was developed.
Methods: We conducted a multicenter survey that collected data on four patients with IOPD, aged 6 to 16 years, who were switched to neoGAA thanks to a compassionate use program, after being treated for an average of 11.5 years with rhGAA. Follow-up data, including biochemical parameters and clinical features, were analyzed to determine clinical outcomes and the safety profile after a mean of 9 months.
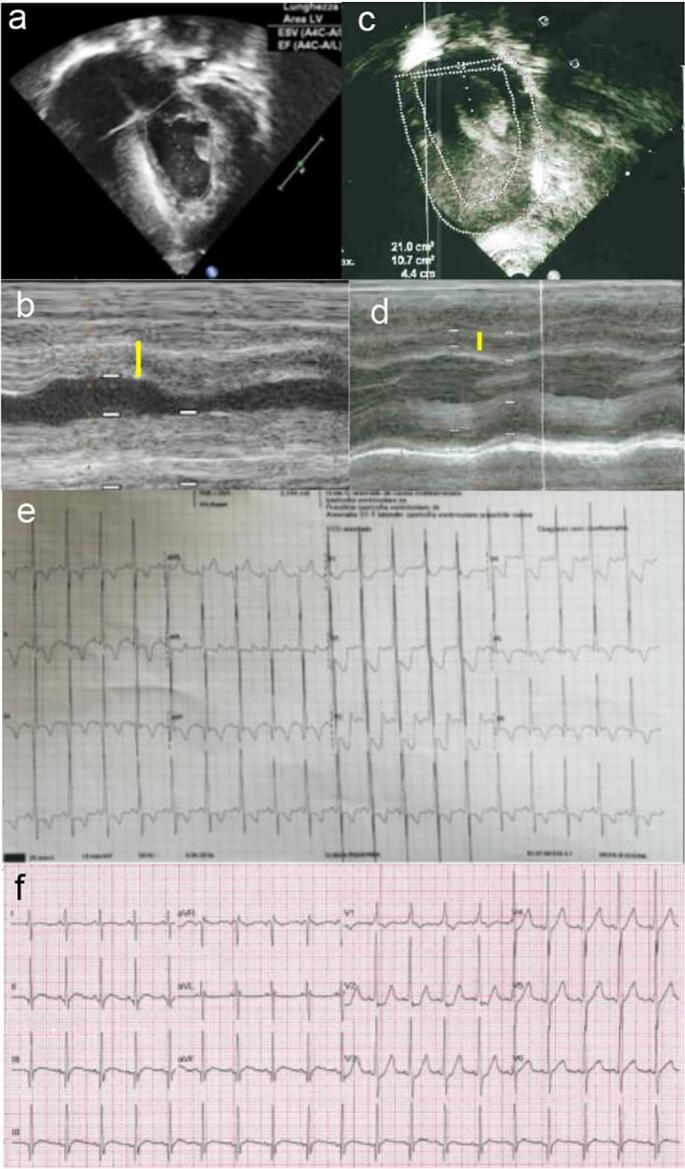
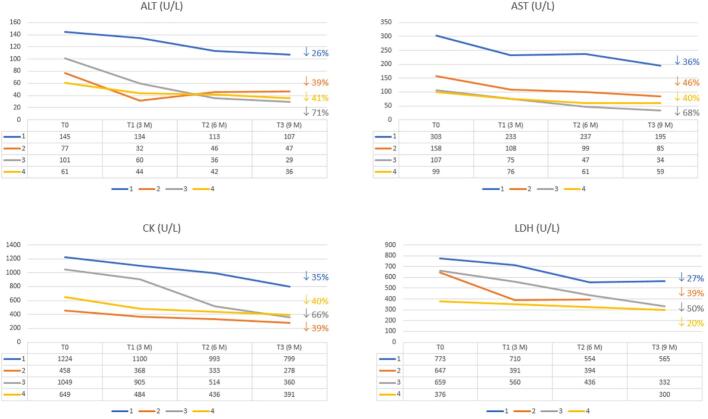
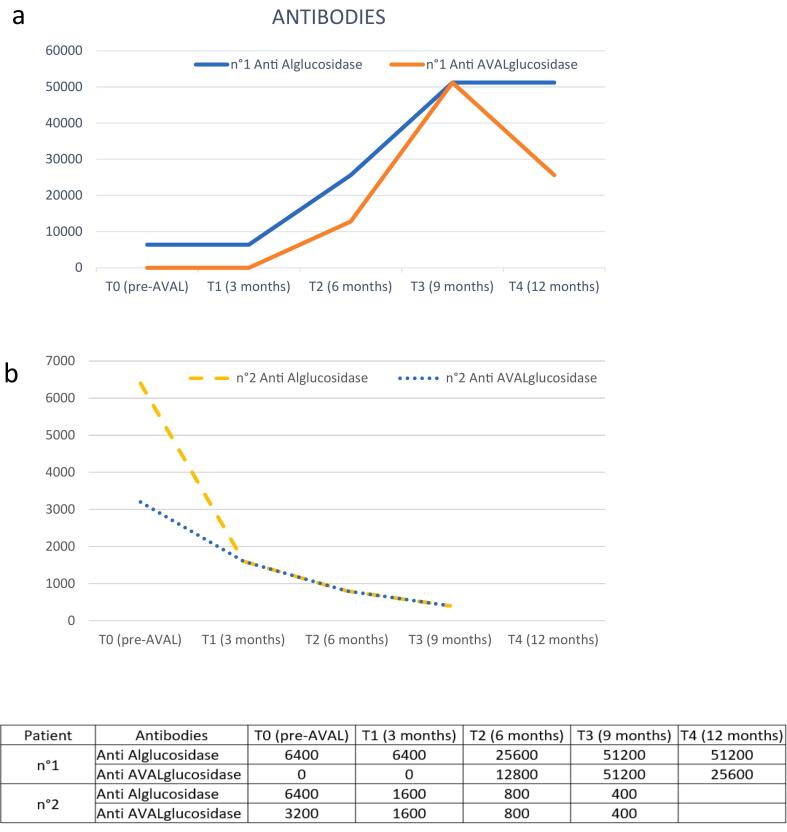
Results: Patients with IOPD who were treated with neoGAA showed a positive change in biomarker levels. Moreover, the clinical picture revealed improved motor performance and cardiac parameters in patients who previously responded poorly.
Conclusion: This study highlights the improved efficacy of neoGAA, as a next generation enzyme replacement therapy, in 4 Italian patients with IOPD. Several clinical parameters showed a positive response to the new formulation suggesting that, if used at diagnosis, neoGAA may result in better outcomes for patients with IOPD.
© 2024 The Authors. Published by Elsevier Inc.
Conflict of interest statement
None.
Figures
References
-
- Reuser A.J., Hirschhorn R., Kroos M.A. McGraw Hill: The Online Metabolic & Molecular Bases of Inherited Disease (OMMBID); 2018. Pompe Disease: Glycogen Storage Disease type II, Acid Alpha-Glucosidase (Acid Maltase) Deficiency.https://ommbid.mhmedical.com
-
- Gungor D., Reuser A.J. How to describe the clinical spectrum in Pompe disease? Am. J. Med. Genet. A. 2013;161 A(2):399–400. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
