

High-resolution epidemiological landscape from ~290,000 SARS-CoV-2 genomes from Denmark
- PMID: 39164246
- PMCID: PMC11335946
- DOI: 10.1038/s41467-024-51371-0
High-resolution epidemiological landscape from ~290,000 SARS-CoV-2 genomes from Denmark
Abstract
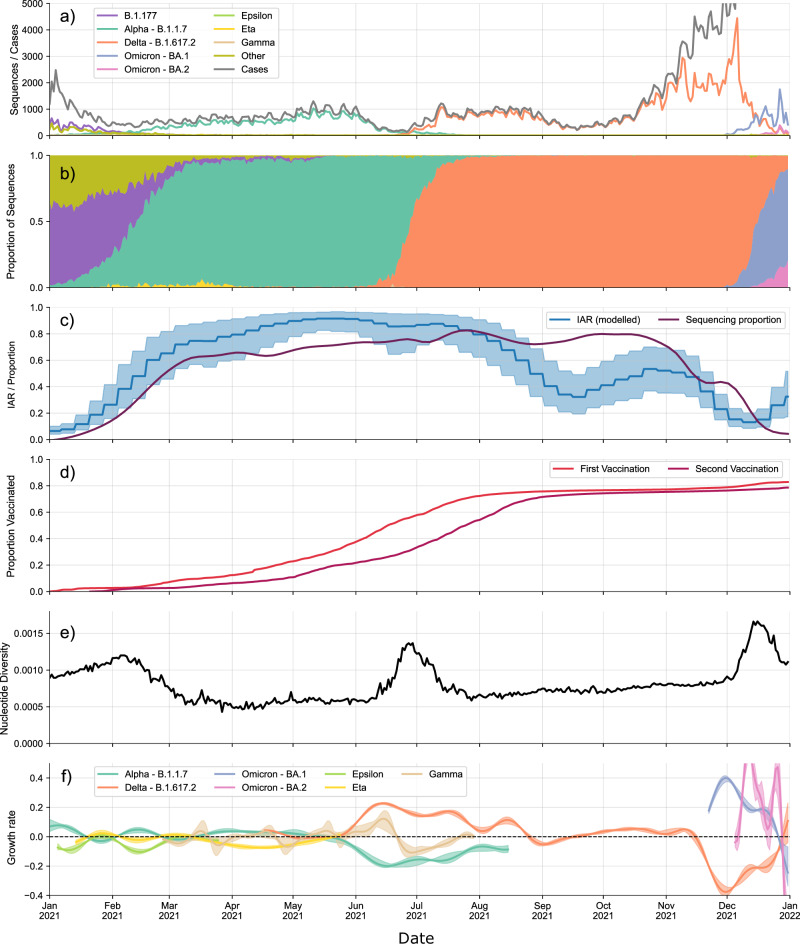
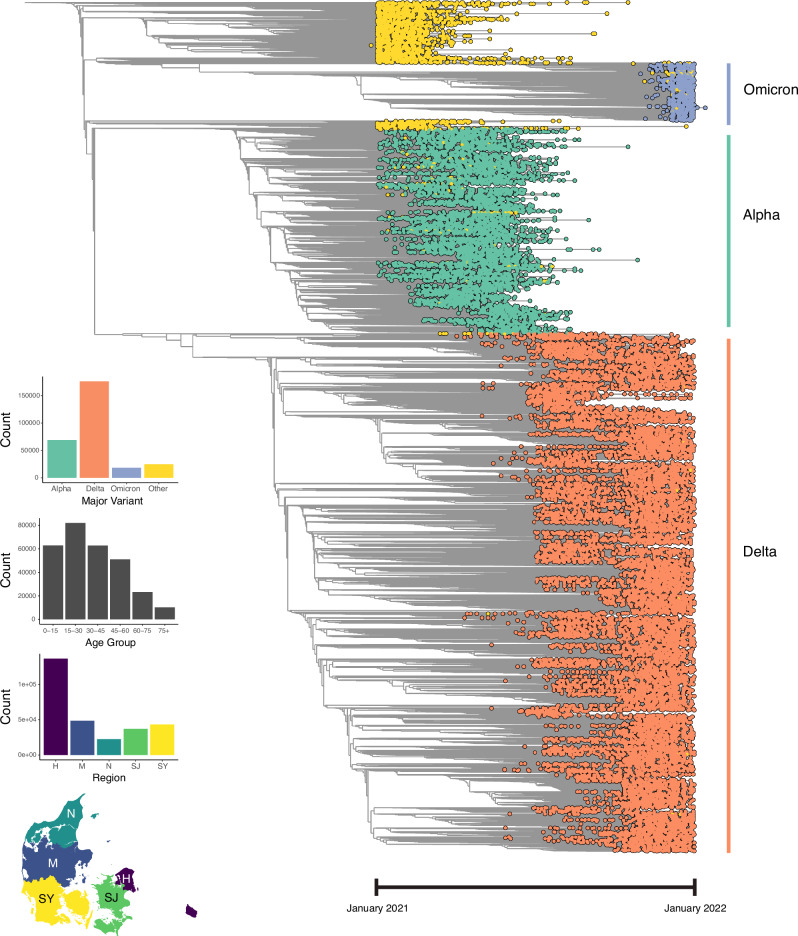
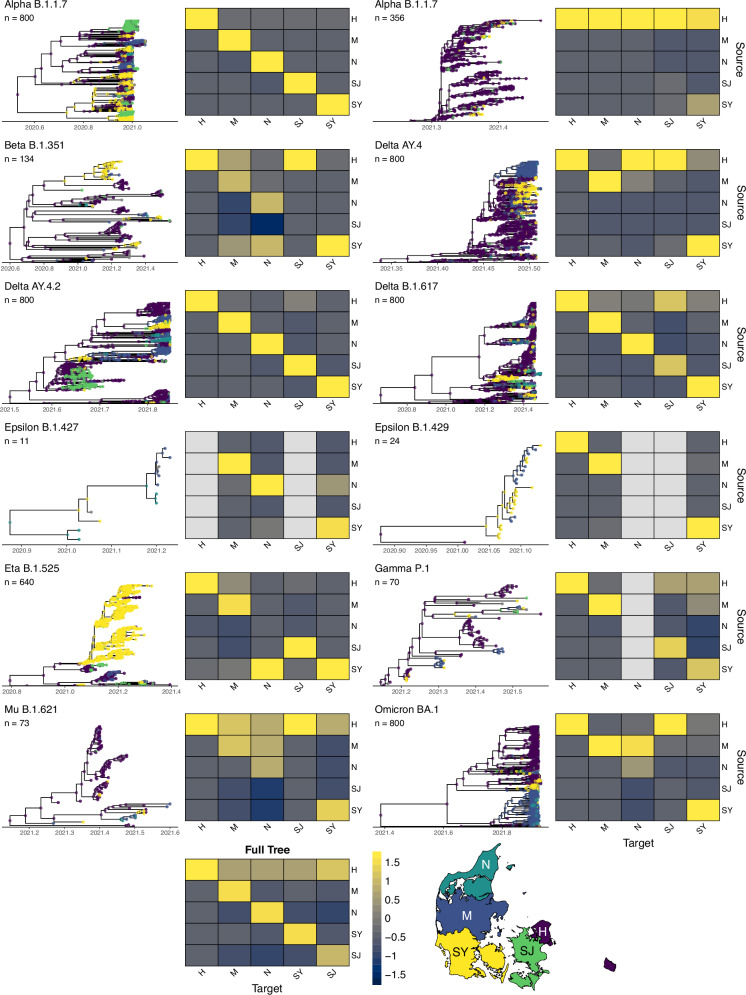
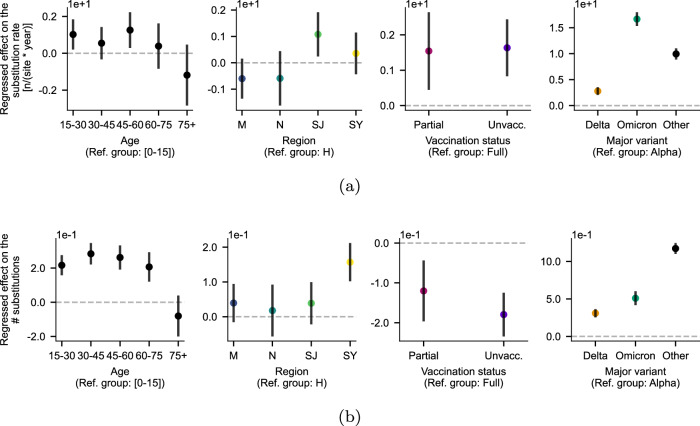
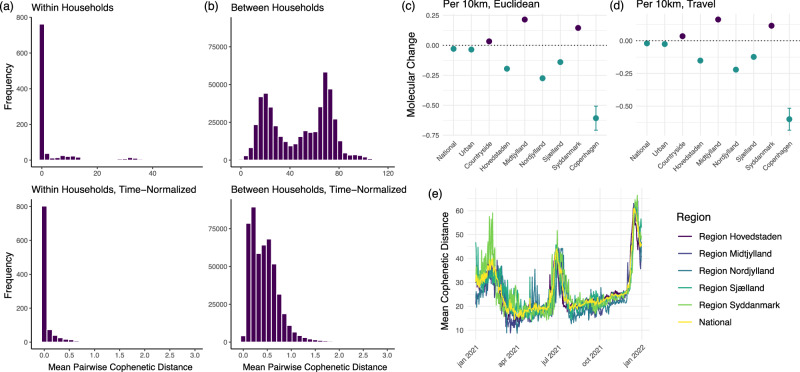
Vast amounts of pathogen genomic, demographic and spatial data are transforming our understanding of SARS-CoV-2 emergence and spread. We examined the drivers of molecular evolution and spread of 291,791 SARS-CoV-2 genomes from Denmark in 2021. With a sequencing rate consistently exceeding 60%, and up to 80% of PCR-positive samples between March and November, the viral genome set is broadly whole-epidemic representative. We identify a consistent rise in viral diversity over time, with notable spikes upon the importation of novel variants (e.g., Delta and Omicron). By linking genomic data with rich individual-level demographic data from national registers, we find that individuals aged < 15 and > 75 years had a lower contribution to molecular change (i.e., branch lengths) compared to other age groups, but similar molecular evolutionary rates, suggesting a lower likelihood of introducing novel variants. Similarly, we find greater molecular change among vaccinated individuals, suggestive of immune evasion. We also observe evidence of transmission in rural areas to follow predictable diffusion processes. Conversely, urban areas are expectedly more complex due to their high mobility, emphasising the role of population structure in driving virus spread. Our analyses highlight the added value of integrating genomic data with detailed demographic and spatial information, particularly in the absence of structured infection surveys.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
