

Generative artificial intelligence performs rudimentary structural biology modeling
- PMID: 39169047
- PMCID: PMC11339285
- DOI: 10.1038/s41598-024-69021-2
Generative artificial intelligence performs rudimentary structural biology modeling
Abstract
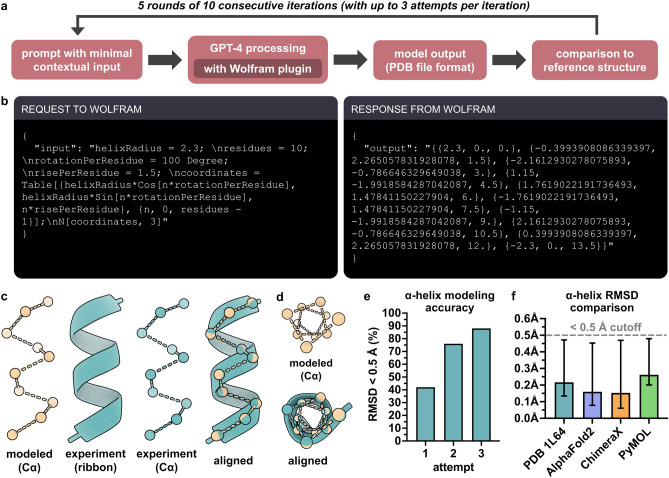
Natural language-based generative artificial intelligence (AI) has become increasingly prevalent in scientific research. Intriguingly, capabilities of generative pre-trained transformer (GPT) language models beyond the scope of natural language tasks have recently been identified. Here we explored how GPT-4 might be able to perform rudimentary structural biology modeling. We prompted GPT-4 to model 3D structures for the 20 standard amino acids and an α-helical polypeptide chain, with the latter incorporating Wolfram mathematical computation. We also used GPT-4 to perform structural interaction analysis between the anti-viral nirmatrelvir and its target, the SARS-CoV-2 main protease. Geometric parameters of the generated structures typically approximated close to experimental references. However, modeling was sporadically error-prone and molecular complexity was not well tolerated. Interaction analysis further revealed the ability of GPT-4 to identify specific amino acid residues involved in ligand binding along with corresponding bond distances. Despite current limitations, we show the current capacity of natural language generative AI to perform basic structural biology modeling and interaction analysis with atomic-scale accuracy.
Keywords: Artificial intelligence; GPT; Language model; Machine learning; Protein modeling; Structural biology.
© 2024. The Author(s).
Conflict of interest statement
A.M.I. is a founder and partner of North Horizon, which is engaged in the development of artificial intelligence-based software. C.M. declares no competing interests. S.K.B. declares no competing interests. M.B.M. declares no competing interests. R.P. is a founder and equity shareholder of PhageNova Bio. R.P. is Chief Scientific Officer and a paid consultant of PhageNova Bio. R.P. is a founder and equity shareholder of MBrace Therapeutics. R.P. serves as a paid consultant for MBrace Therapeutics. R.P. has Sponsored Research Agreements (SRAs) in place with PhageNova Bio and with MBrace Therapeutics. These arrangements are managed in accordance with the established institutional conflict-of-interest policies of Rutgers, The State University of New Jersey. This study falls outside of the scope of these SRAs. W.A. is a founder and equity shareholder of PhageNova Bio. W.A. is a founder and equity shareholder of MBrace Therapeutics. W.A. serves as a paid consultant for MBrace Therapeutics. W.A. has Sponsored Research Agreements (SRAs) in place with PhageNova Bio and with MBrace Therapeutics. These arrangements are managed in accordance with the established institutional conflict-of-interest policies of Rutgers, The State University of New Jersey. This study falls outside of the scope of these SRAs.
Figures


Update of
-
Generative artificial intelligence performs rudimentary structural biology modeling.bioRxiv [Preprint]. 2024 May 13:2024.01.10.575113. doi: 10.1101/2024.01.10.575113. bioRxiv. 2024. Update in: Sci Rep. 2024 Aug 21;14(1):19372. doi: 10.1038/s41598-024-69021-2. PMID: 38293060 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
