

NAD+ precursors prolong survival and improve cardiac phenotypes in a mouse model of Friedreich's Ataxia
- PMID: 39171530
- PMCID: PMC11343603
- DOI: 10.1172/jci.insight.177152
NAD+ precursors prolong survival and improve cardiac phenotypes in a mouse model of Friedreich's Ataxia
Abstract
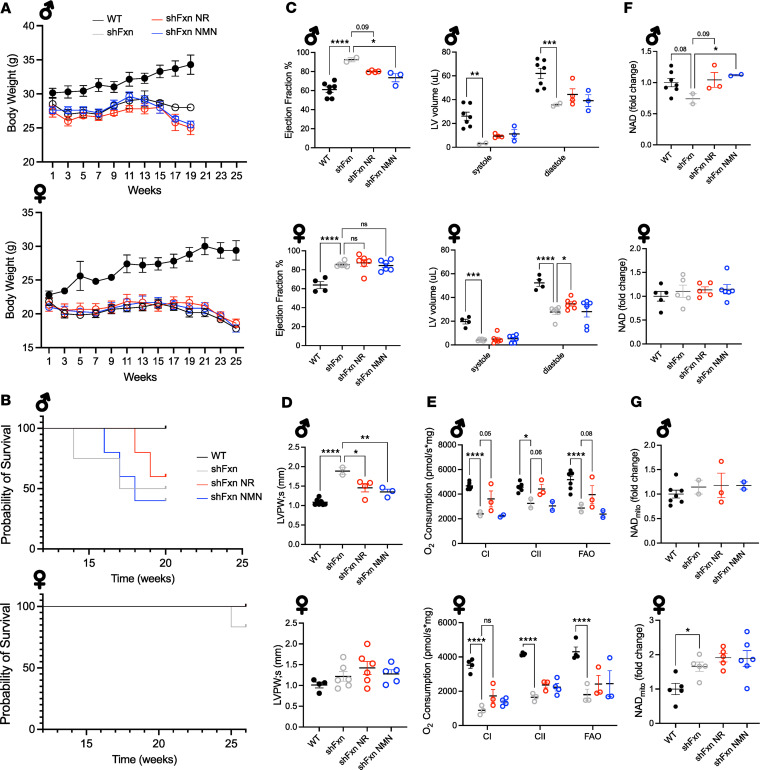
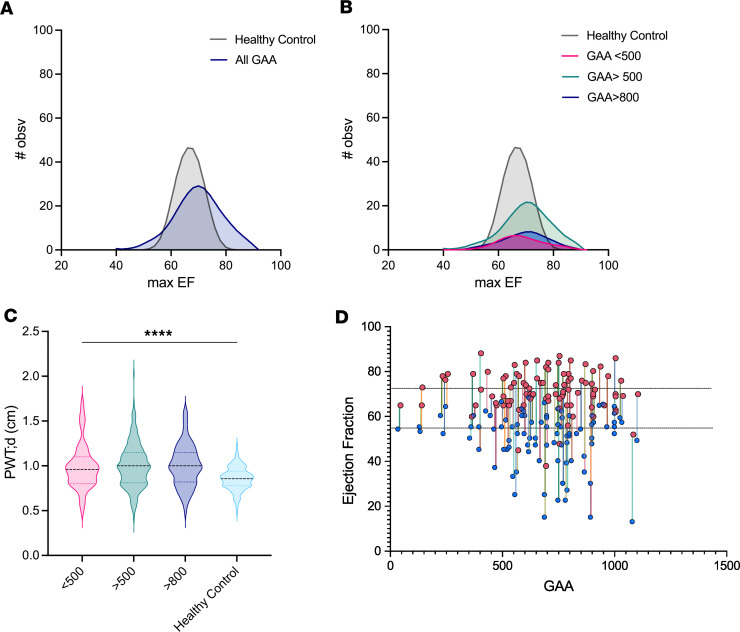
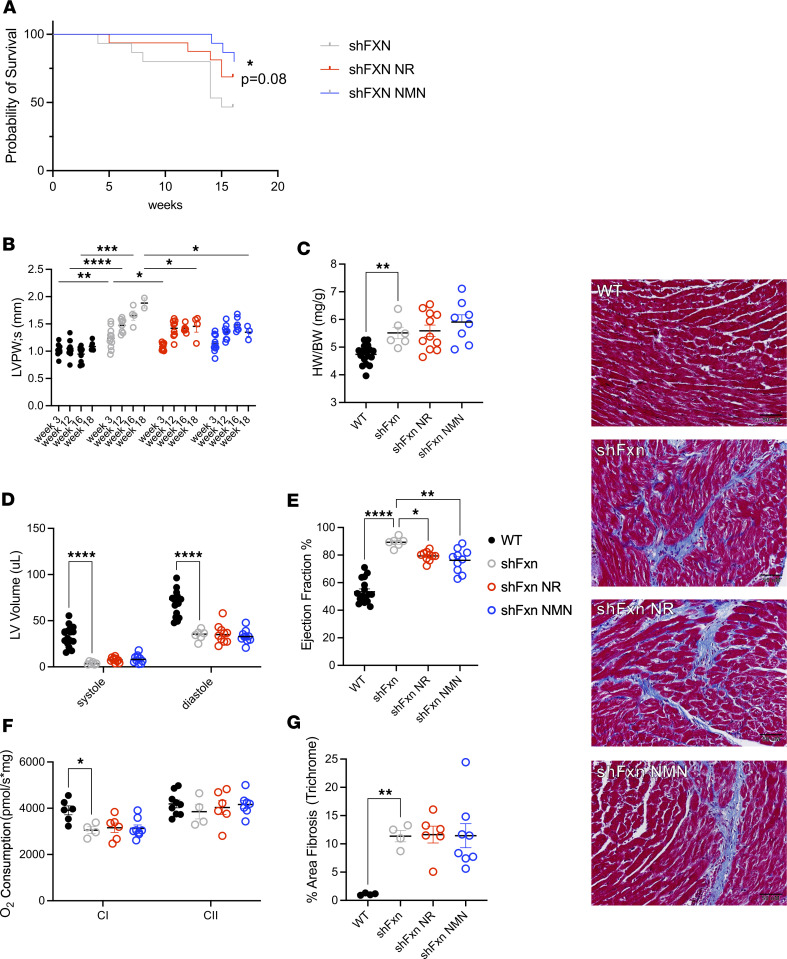
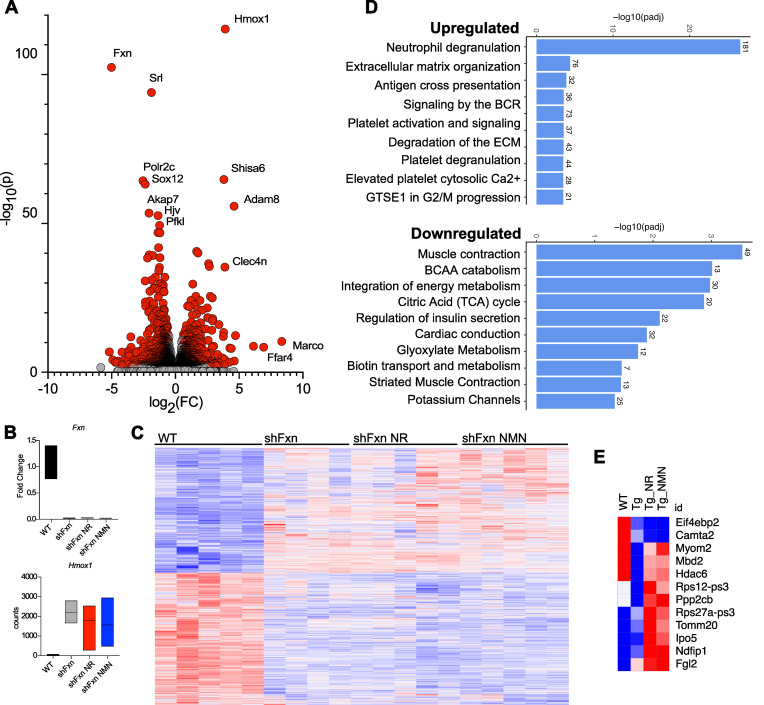
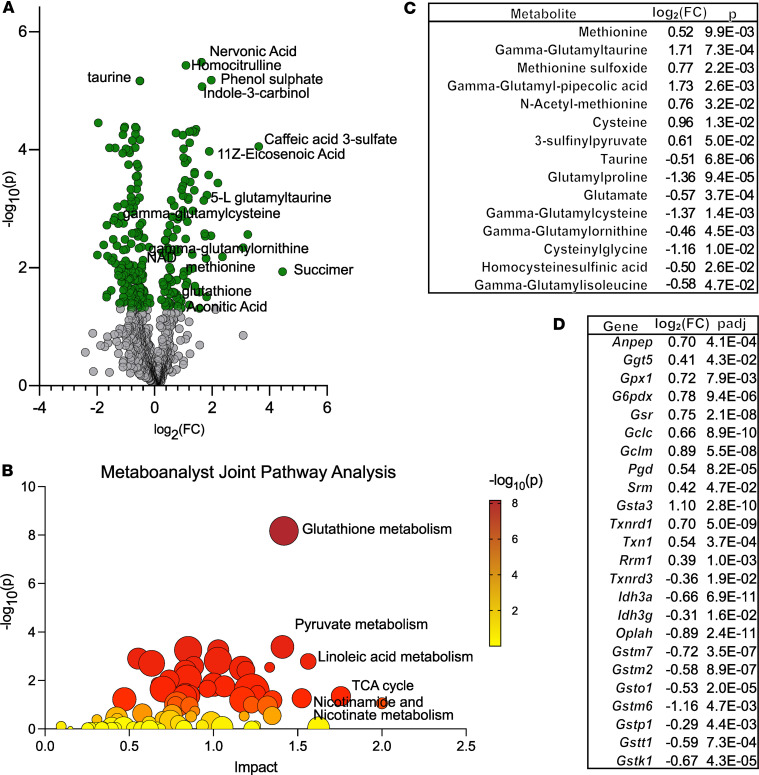
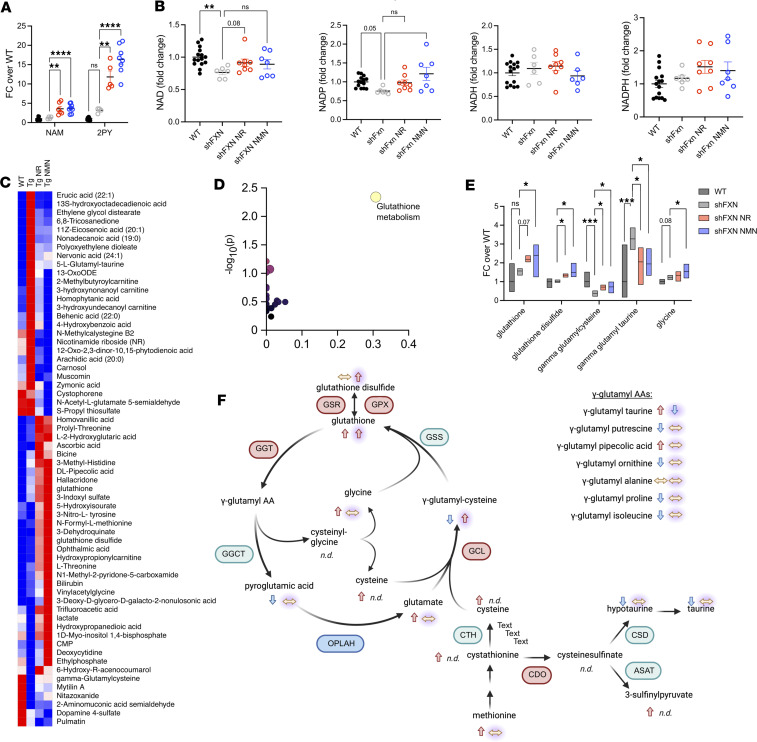
Friedreich's ataxia (FRDA) is a progressive disorder caused by insufficient expression of frataxin, which plays a critical role in assembly of iron-sulfur centers in mitochondria. Individuals are cognitively normal but display a loss of motor coordination and cardiac abnormalities. Many ultimately develop heart failure. Administration of nicotinamide adenine dinucleotide-positive (NAD+) precursors has shown promise in human mitochondrial myopathy and rodent models of heart failure, including mice lacking frataxin in cardiomyocytes. We studied mice with systemic knockdown of frataxin (shFxn), which display motor deficits and early mortality with cardiac hypertrophy. Hearts in these mice do not "fail" per se but become hyperdynamic with small chamber sizes. Data from an ongoing natural history study indicate that hyperdynamic hearts are observed in young individuals with FRDA, suggesting that the mouse model could reflect early pathology. Administering nicotinamide mononucleotide or riboside to shFxn mice increases survival, modestly improves cardiac hypertrophy, and limits increases in ejection fraction. Mechanistically, most of the transcriptional and metabolic changes induced by frataxin knockdown are insensitive to NAD+ precursor administration, but glutathione levels are increased, suggesting improved antioxidant capacity. Overall, our findings indicate that NAD+ precursors are modestly cardioprotective in this model of FRDA and warrant further investigation.
Keywords: Cardiology; Metabolism; Mitochondria; Neurological disorders.
Conflict of interest statement
Figures
References
-
- Bidichandani SI, et al. In: Adam MP, Feldman J, Mirzaa GM, et al., eds. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1998.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
