

Primary unilateral macronodular adrenal hyperplasia with concomitant glucocorticoid and androgen excess and KDM1A inactivation
- PMID: 39171930
- PMCID: PMC11378072
- DOI: 10.1093/ejendo/lvae106
Primary unilateral macronodular adrenal hyperplasia with concomitant glucocorticoid and androgen excess and KDM1A inactivation
Abstract
Background: Primary bilateral macronodular adrenal hyperplasia (PBMAH) is a rare cause of Cushing's syndrome. Individuals with PBMAH and glucose-dependent insulinotropic polypeptide (GIP)-dependent Cushing's syndrome due to ectopic expression of the GIP receptor (GIPR) typically harbor inactivating KDM1A sequence variants. Primary unilateral macronodular adrenal hyperplasia (PUMAH) with concomitant glucocorticoid and androgen excess has never been encountered or studied.
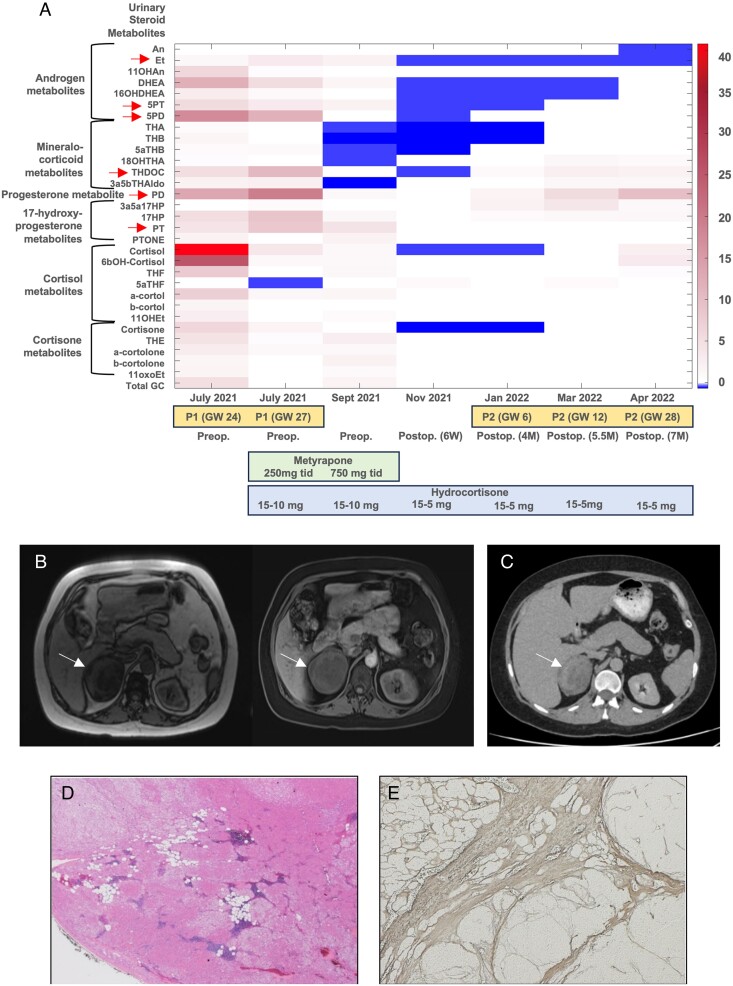
Methods: We investigated a woman with a large, heterogeneous adrenal mass and severe adrenocorticotropic hormone-independent glucocorticoid and androgen excess, a biochemical presentation typically suggestive of adrenocortical carcinoma. The patient presented during pregnancy (22nd week of gestation) and reported an 18-month history of oligomenorrhea, hirsutism, and weight gain. We undertook an exploratory study with detailed histopathological and genetic analysis of the resected adrenal mass and leukocyte DNA collected from the patient and her parents.
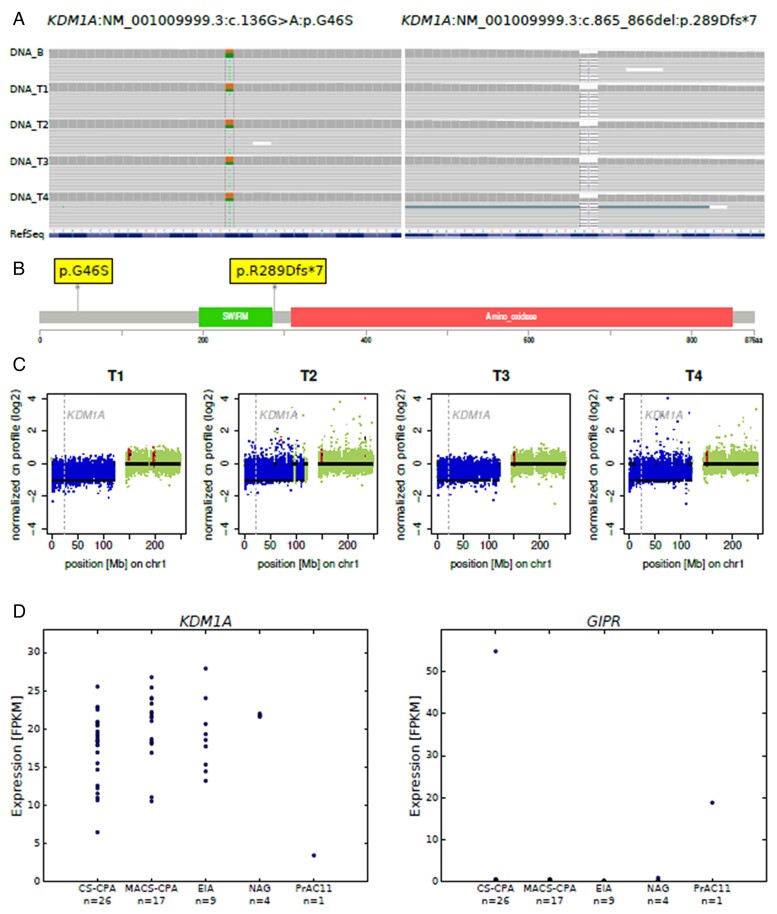
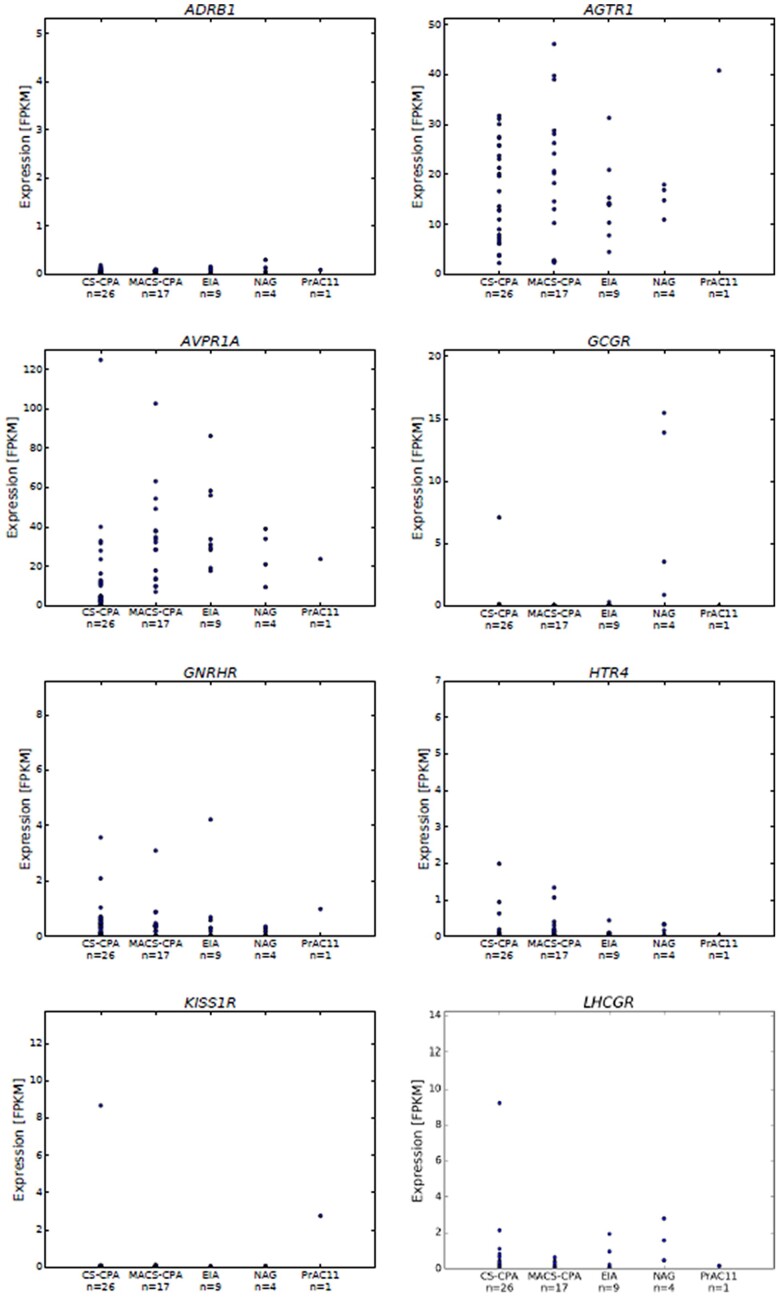
Results: Histopathology revealed benign macronodular adrenal hyperplasia. Imaging showed a persistently normal contralateral adrenal gland. Whole-exome sequencing of 4 representative nodules detected KDM1A germline variants, benign NM_001009999.3:c.136G > A:p.G46S, and likely pathogenic NM_001009999.3:exon6:c.865_866del:p.R289Dfs*7. Copy number variation analysis demonstrated an additional somatic loss of the KDM1A wild-type allele on chromosome 1p36.12 in all nodules. RNA sequencing of a representative nodule showed low/absent KDM1A expression and increased GIPR expression compared with 52 unilateral sporadic adenomas and 4 normal adrenal glands. Luteinizing hormone/chorionic gonadotropin receptor expression was normal. Sanger sequencing confirmed heterozygous KDM1A variants in both parents (father: p.R289Dfs*7 and mother: p.G46S) who showed no clinical features suggestive of glucocorticoid or androgen excess.
Conclusions: We investigated the first PUMAH associated with severe Cushing's syndrome and concomitant androgen excess, suggesting pathogenic mechanisms involving KDM1A.
Keywords: KDM1A; MC2R; Cushing's syndrome; GIP receptor; PBMAH; PUMAH; adrenocortical tumor; androgen excess.
© The Author(s) 2024. Published by Oxford University Press on behalf of European Society of Endocrinology.
Conflict of interest statement
Conflict of interest: none declared.
Figures
Similar articles
-
Loss of KDM1A in GIP-dependent primary bilateral macronodular adrenal hyperplasia with Cushing's syndrome: a multicentre, retrospective, cohort study.Lancet Diabetes Endocrinol. 2021 Dec;9(12):813-824. doi: 10.1016/S2213-8587(21)00236-9. Epub 2021 Oct 13. Lancet Diabetes Endocrinol. 2021. PMID: 34655521
-
Extensive expertise in endocrinology: glucose-dependent insulinotropic peptide-dependent Cushing's syndrome.Eur J Endocrinol. 2023 Mar 2;188(3):R56-R72. doi: 10.1093/ejendo/lvad026. Eur J Endocrinol. 2023. PMID: 36857084
-
Clinical, Pathophysiologic, Genetic, and Therapeutic Progress in Primary Bilateral Macronodular Adrenal Hyperplasia.Endocr Rev. 2023 Jul 11;44(4):567-628. doi: 10.1210/endrev/bnac034. Endocr Rev. 2023. PMID: 36548967
-
From the First Case Reports to KDM1A Identification: 35 Years of Food (GIP)-Dependent Cushing's Syndrome.Exp Clin Endocrinol Diabetes. 2024 Dec;132(12):697-704. doi: 10.1055/a-2359-8051. Epub 2024 Jul 26. Exp Clin Endocrinol Diabetes. 2024. PMID: 39059410 Review.
-
Primary bilateral macronodular adrenal hyperplasia: definitely a genetic disease.Nat Rev Endocrinol. 2022 Nov;18(11):699-711. doi: 10.1038/s41574-022-00718-y. Epub 2022 Aug 3. Nat Rev Endocrinol. 2022. PMID: 35922573 Review.
Cited by
-
Sexual dimorphism in benign adrenocortical tumours.Eur J Endocrinol. 2025 Apr 30;192(5):R1-R12. doi: 10.1093/ejendo/lvaf088. Eur J Endocrinol. 2025. PMID: 40296186 Free PMC article. Review.
References
-
- Fassnacht M, Tsagarakis S, Terzolo M, et al. European Society of Endocrinology clinical practice guidelines on the management of adrenal incidentalomas, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol. 2023;189(1):G1–G42. 10.1093/ejendo/lvad066 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
