

Enhancing neurogenesis after traumatic brain injury: The role of adenosine kinase inhibition in promoting neuronal survival and differentiation
- PMID: 39173898
- PMCID: PMC12042129
- DOI: 10.1016/j.expneurol.2024.114930
Enhancing neurogenesis after traumatic brain injury: The role of adenosine kinase inhibition in promoting neuronal survival and differentiation
Abstract
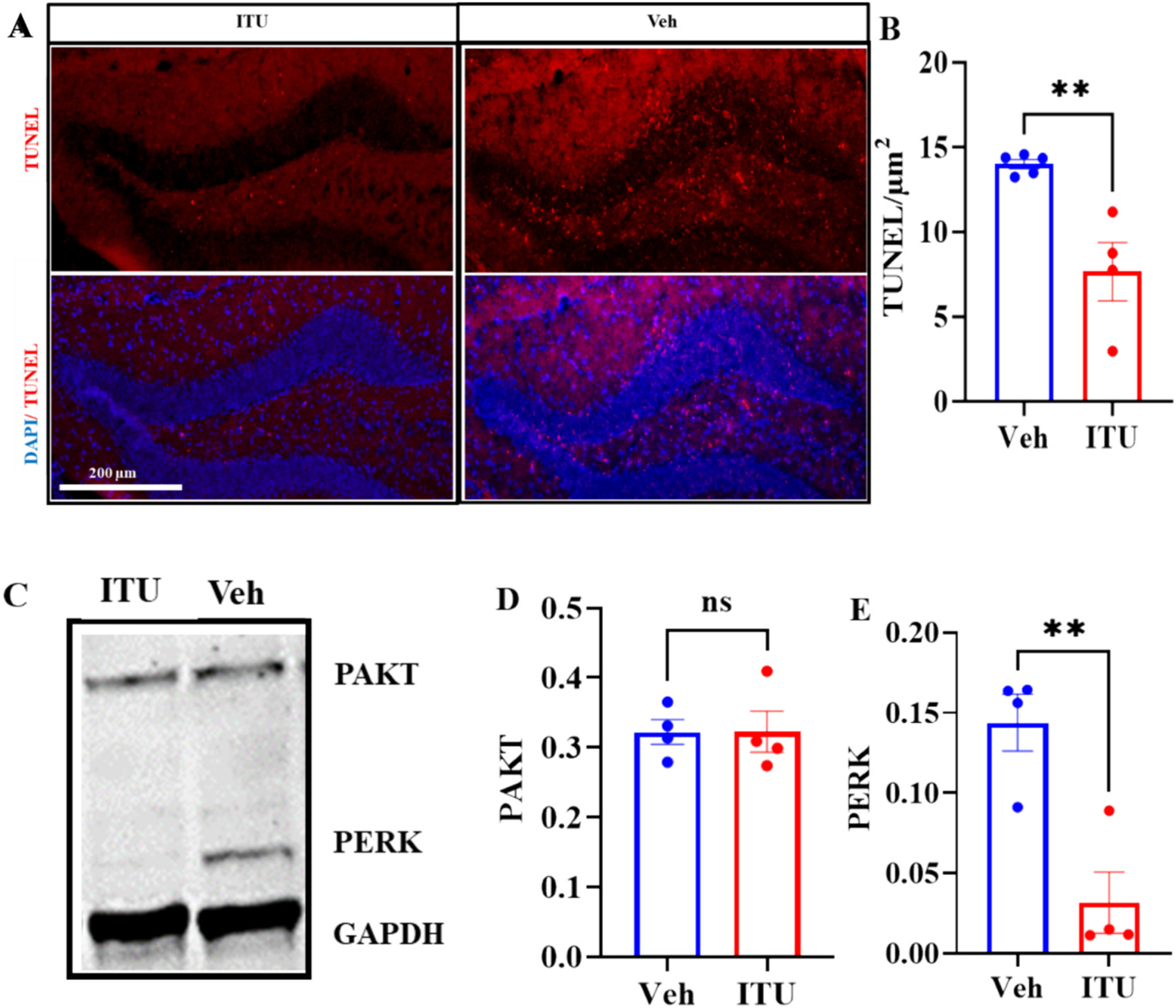
Traumatic brain injury (TBI) presents a significant public health challenge, necessitating innovative interventions for effective treatment. Recent studies have challenged conventional perspectives on neurogenesis, unveiling endogenous repair mechanisms within the adult brain following injury. However, the intricate mechanisms governing post-TBI neurogenesis remain unclear. The microenvironment of an injured brain, characterized by astrogliosis, neuroinflammation, and excessive cell death, significantly influences the fate of newly generated neurons. Adenosine kinase (ADK), the key metabolic regulator of adenosine, emerges as a crucial factor in brain development and cell proliferation after TBI. This study investigates the hypothesis that targeting ADK could enhance brain repair, promote neuronal survival, and facilitate differentiation. In a TBI model induced by controlled cortical impact, C57BL/6 male mice received intraperitoneal injections of the small molecule ADK inhibitor 5-iodotubercidin (ITU) for three days following TBI. To trace the fate of TBI-associated proliferative cells, animals received intraperitoneal injections of BrdU for seven days, beginning immediately after TBI. Our results show that ADK inhibition by ITU improved brain repair 14 days after injury as evidenced by a diminished injury size. Additionally, the number of mature neurons generated after TBI was increased in ITU-treated mice. Remarkably, the TBI-associated pathological events including astrogliosis, neuroinflammation, and cell death were arrested in ITU-treated mice. Finally, ADK inhibition modulated cell death by regulating the PERK signaling pathway. Together, these findings demonstrate a novel therapeutic approach to target multiple pathological mechanisms involved in TBI. This research contributes valuable insights into the intricate molecular mechanisms underlying neurogenesis and gliosis after TBT.
Keywords: Adenosine kinase; Astroglia; Microglia; Neurogenesis; Regeneration; Traumatic brain injury; hippocampus.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare that they have no conflict of interest.
Figures
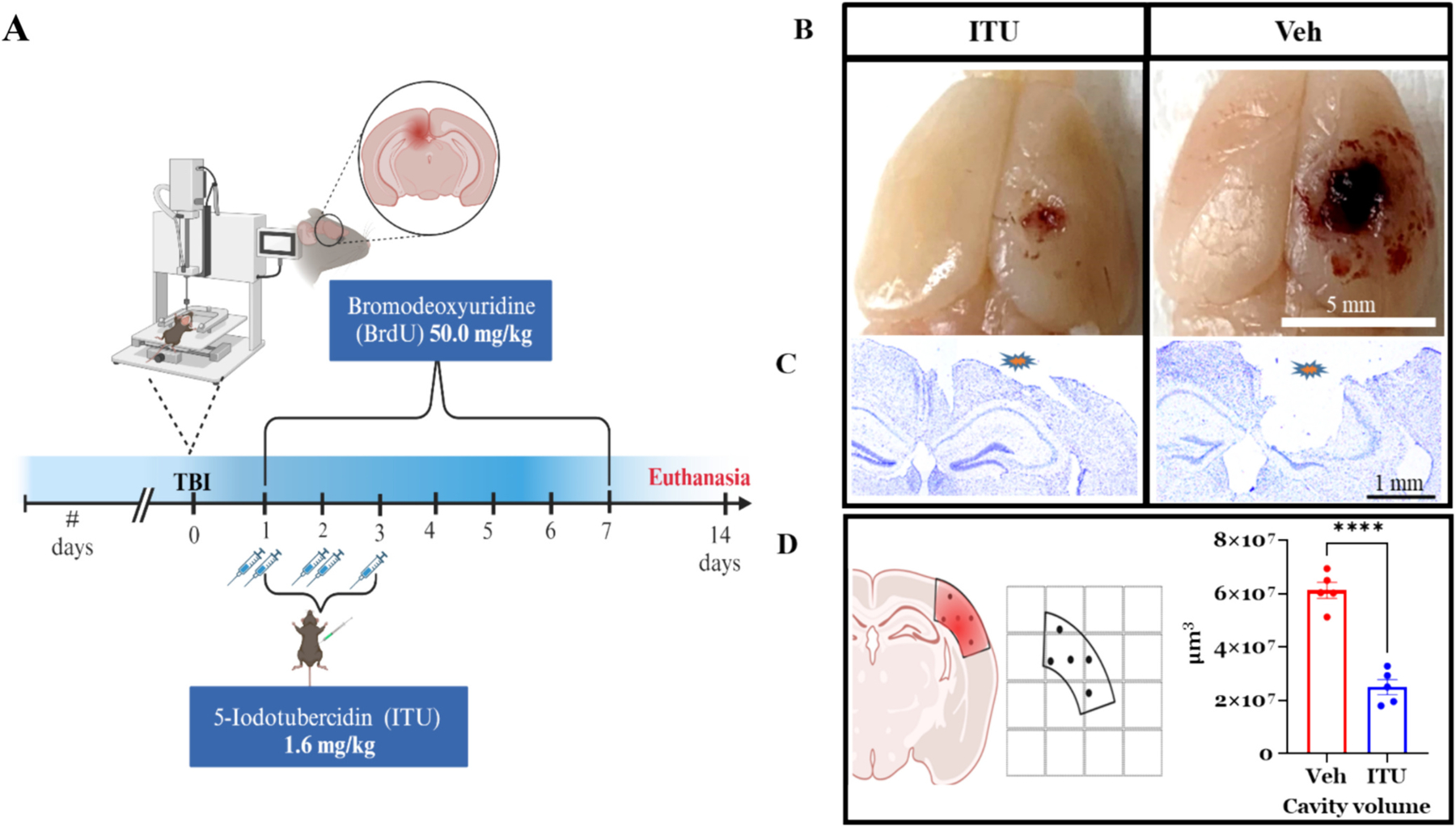
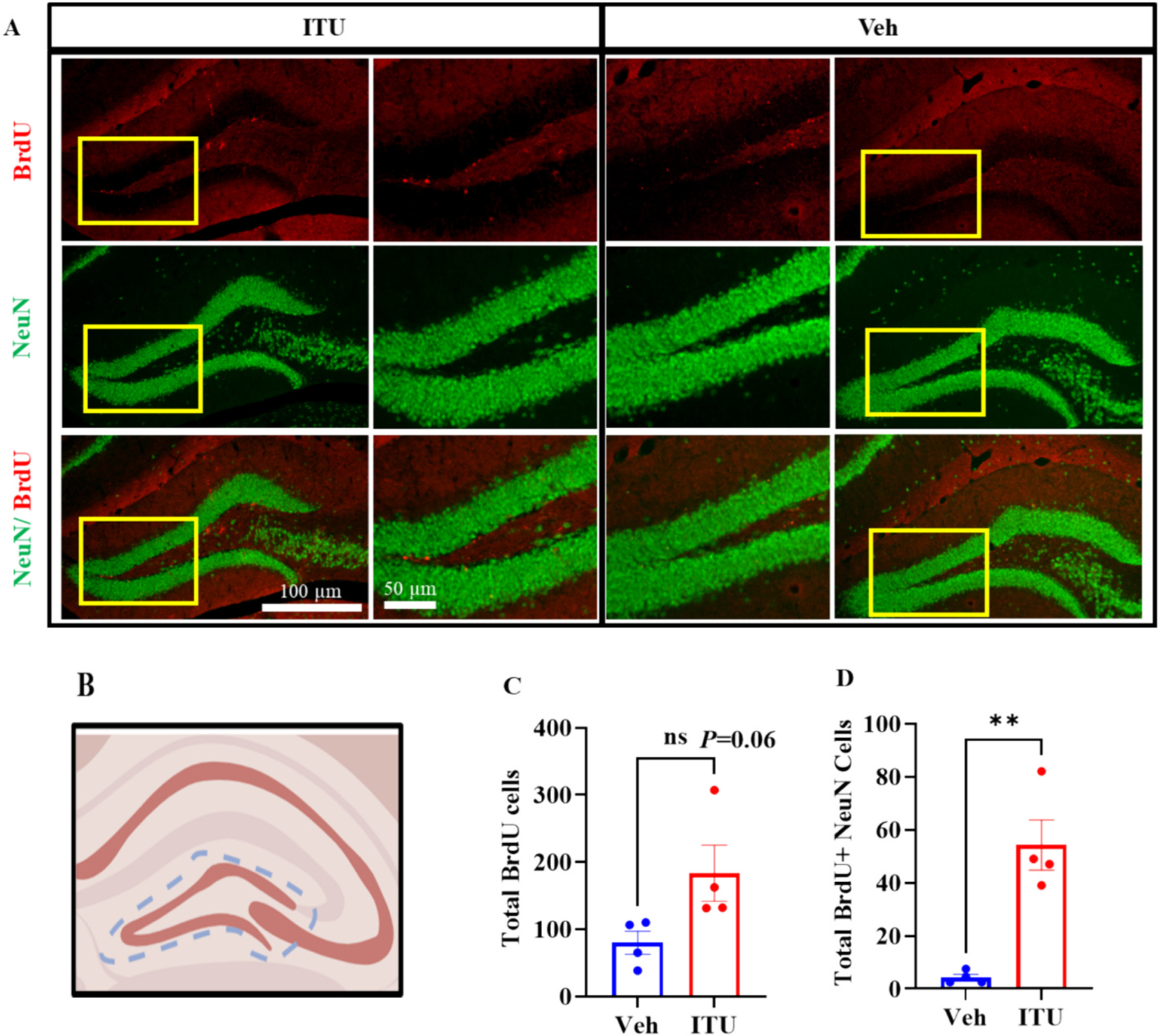
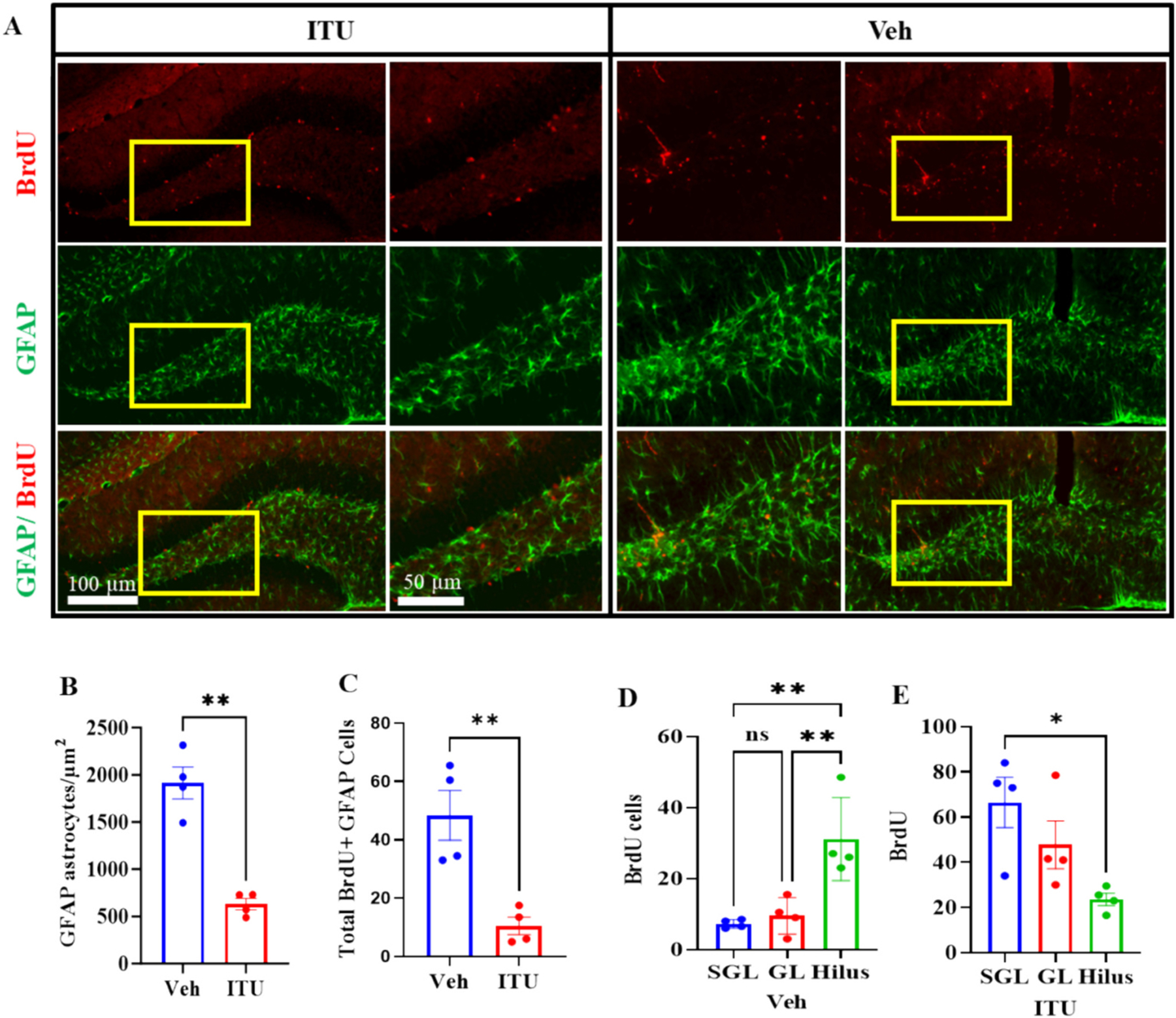
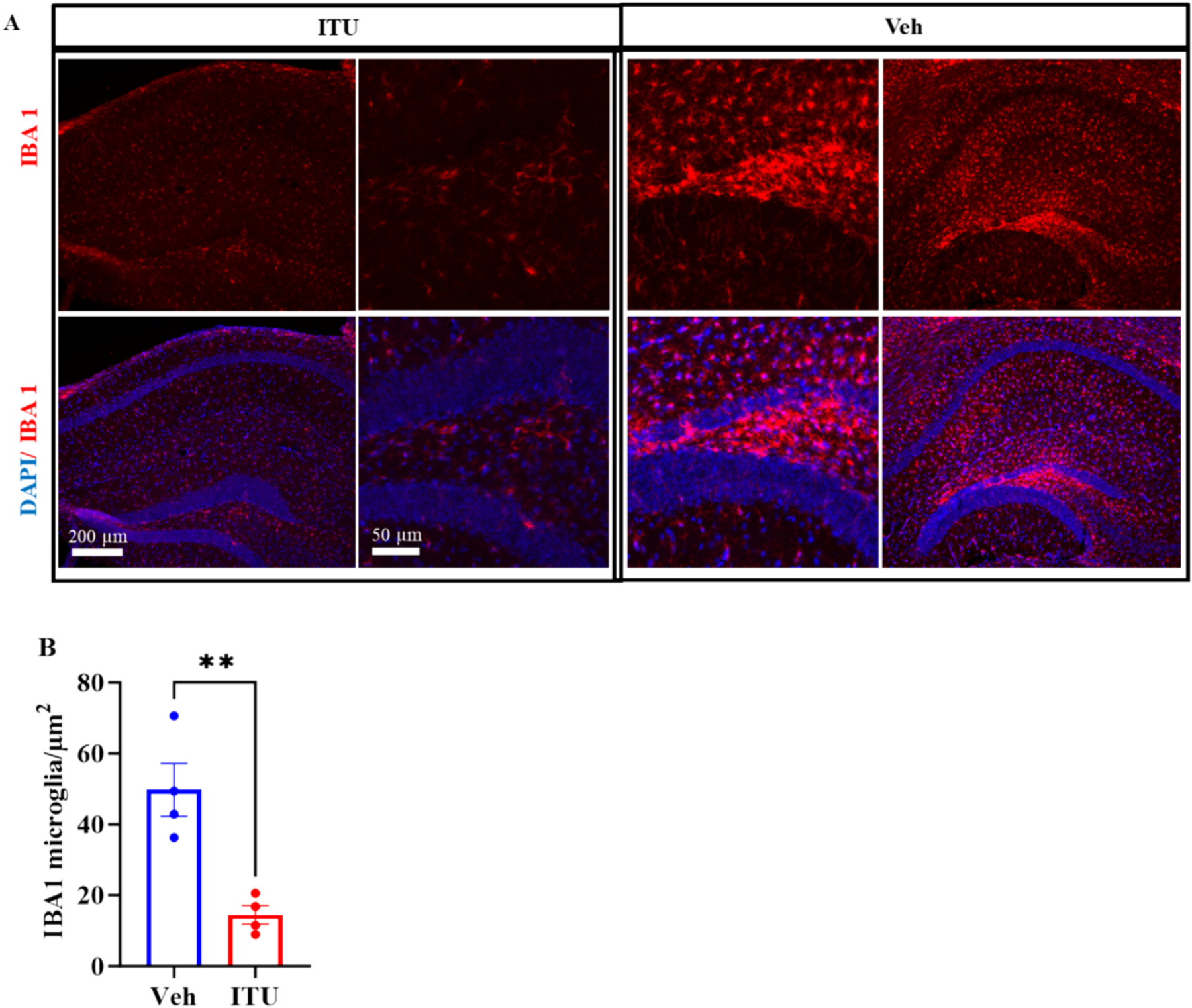
Similar articles
-
Adenosine kinase facilitated astrogliosis-induced cortical neuronal death in traumatic brain injury.J Mol Histol. 2016 Jun;47(3):259-71. doi: 10.1007/s10735-016-9670-7. Epub 2016 Mar 16. J Mol Histol. 2016. PMID: 26983602
-
Adenosine kinase inhibition promotes proliferation of neural stem cells after traumatic brain injury.Brain Commun. 2020;2(1):fcaa017. doi: 10.1093/braincomms/fcaa017. Epub 2020 Feb 20. Brain Commun. 2020. PMID: 32322821 Free PMC article.
-
Transient use of a systemic adenosine kinase inhibitor attenuates epilepsy development in mice.Epilepsia. 2019 Apr;60(4):615-625. doi: 10.1111/epi.14674. Epub 2019 Feb 27. Epilepsia. 2019. PMID: 30815855 Free PMC article.
-
Neurogenesis after traumatic brain injury - The complex role of HMGB1 and neuroinflammation.Neuropharmacology. 2021 Feb 1;183:108400. doi: 10.1016/j.neuropharm.2020.108400. Epub 2020 Nov 13. Neuropharmacology. 2021. PMID: 33189765 Review.
-
IGF-1/IGF-R Signaling in Traumatic Brain Injury: Impact on Cell Survival, Neurogenesis, and Behavioral Outcome.In: Kobeissy FH, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton (FL): CRC Press/Taylor & Francis; 2015. Chapter 7. In: Kobeissy FH, editor. Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton (FL): CRC Press/Taylor & Francis; 2015. Chapter 7. PMID: 26269893 Free Books & Documents. Review.
Cited by
-
Modulation of adult hippocampal neurogenesis by interleukin 1 signaling.Neurobiol Sleep Circadian Rhythms. 2025 Apr 25;18(Suppl):100123. doi: 10.1016/j.nbscr.2025.100123. eCollection 2025 May. Neurobiol Sleep Circadian Rhythms. 2025. PMID: 40703575 Free PMC article.
References
-
- Ajizian SJ, English BK, Meals EA, 1999. Specific inhibitors of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways block inducible nitric oxide synthase and tumor necrosis factor accumulation in murine macrophages stimulated with lipopolysaccharide and interferon-gamma. J. Infect. Dis 179 (4), 939–944. 10.1086/314659. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
