

Periodic paralysis
- PMID: 39174253
- PMCID: PMC11556526
- DOI: 10.1016/B978-0-323-90820-7.00002-1
Periodic paralysis
Abstract
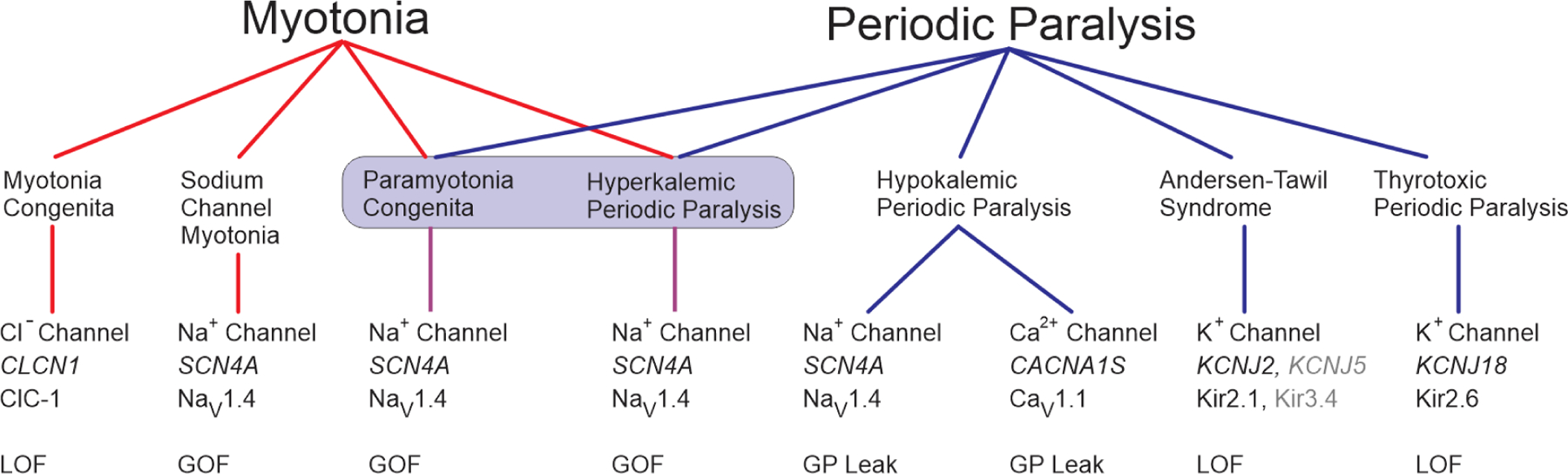
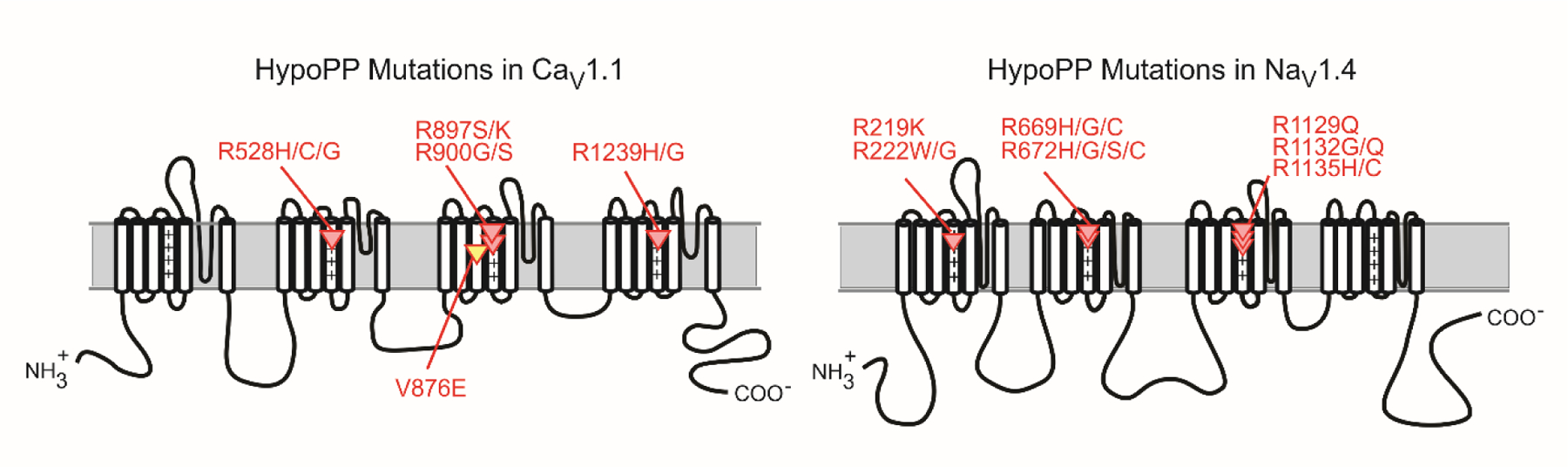
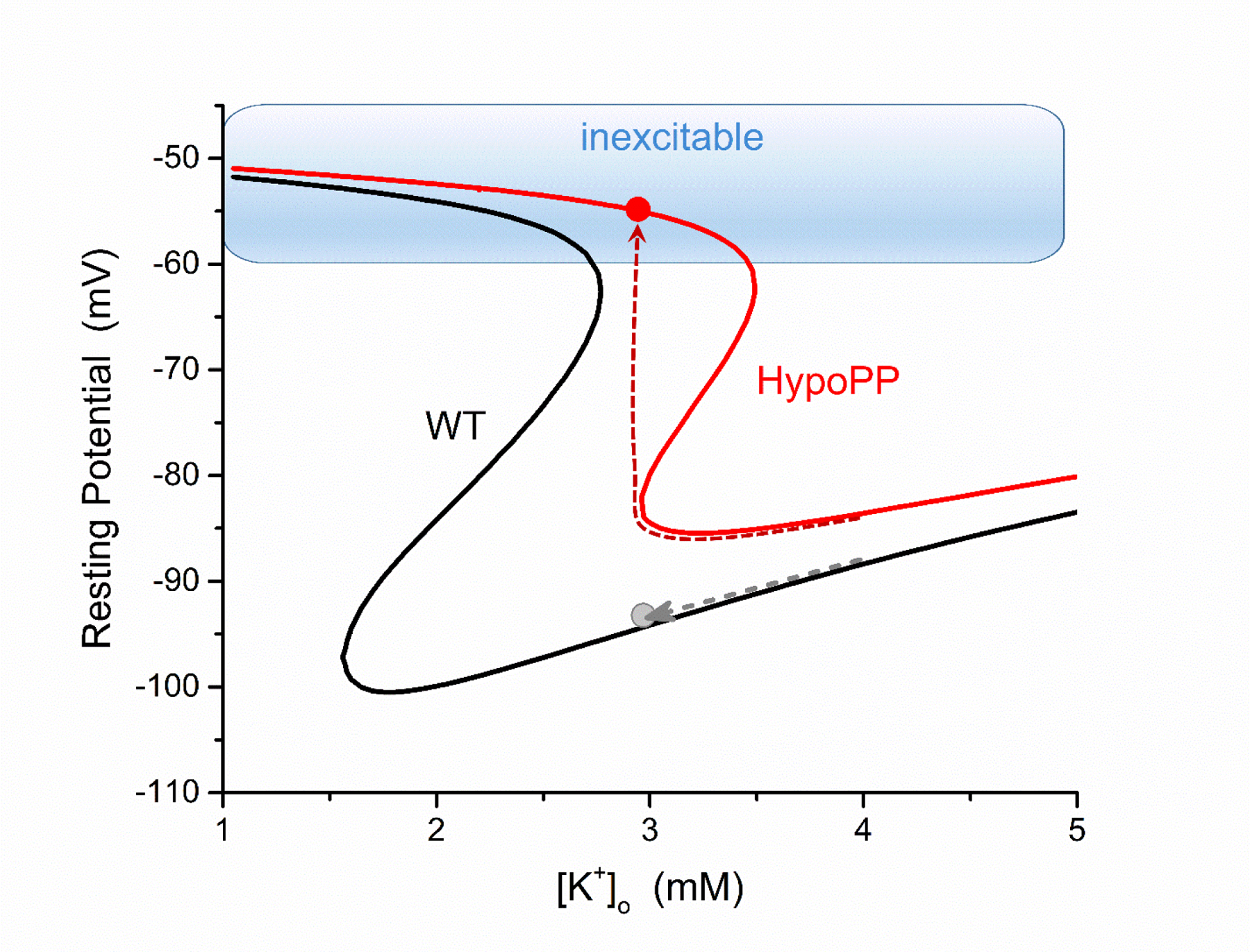
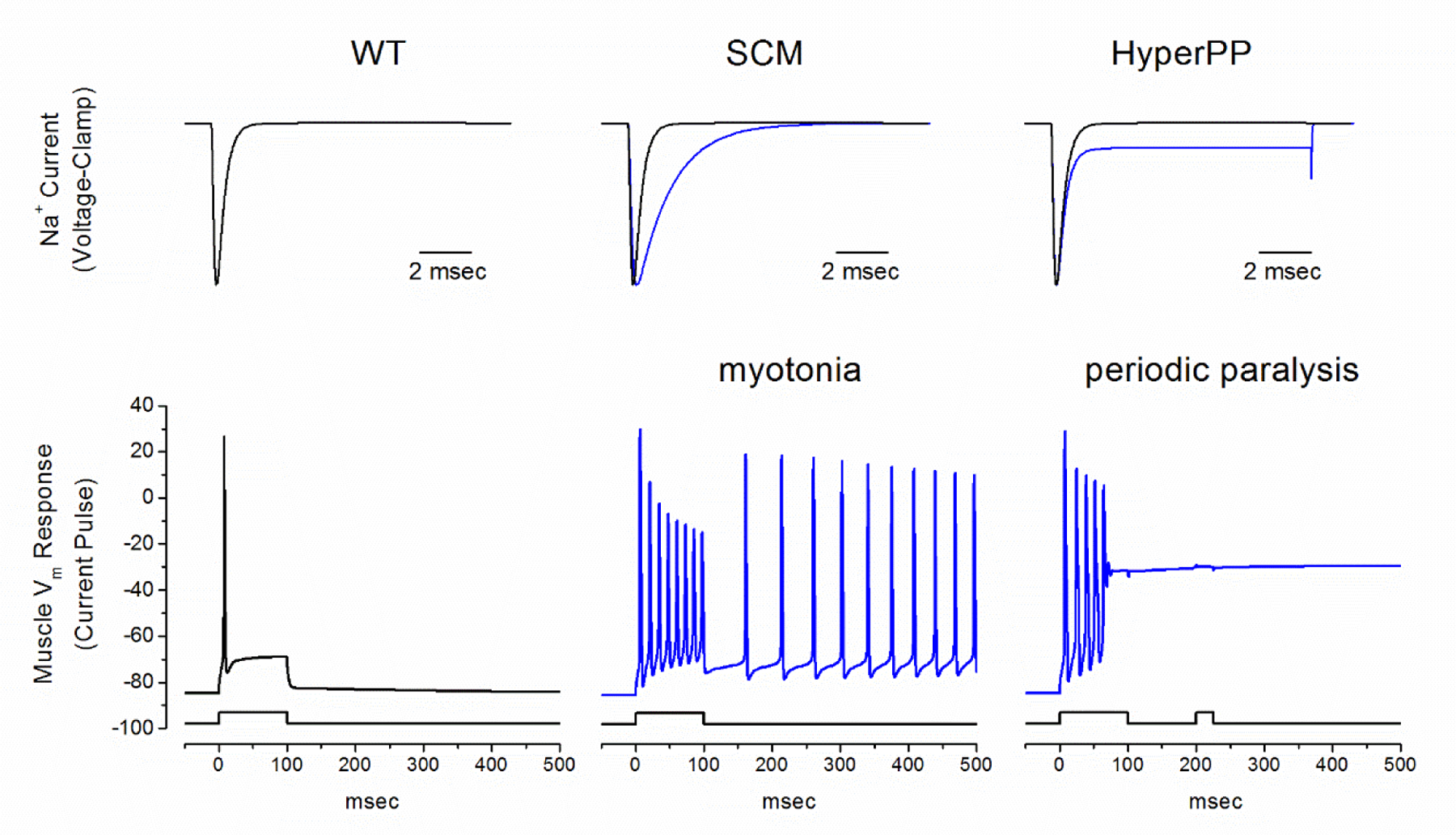
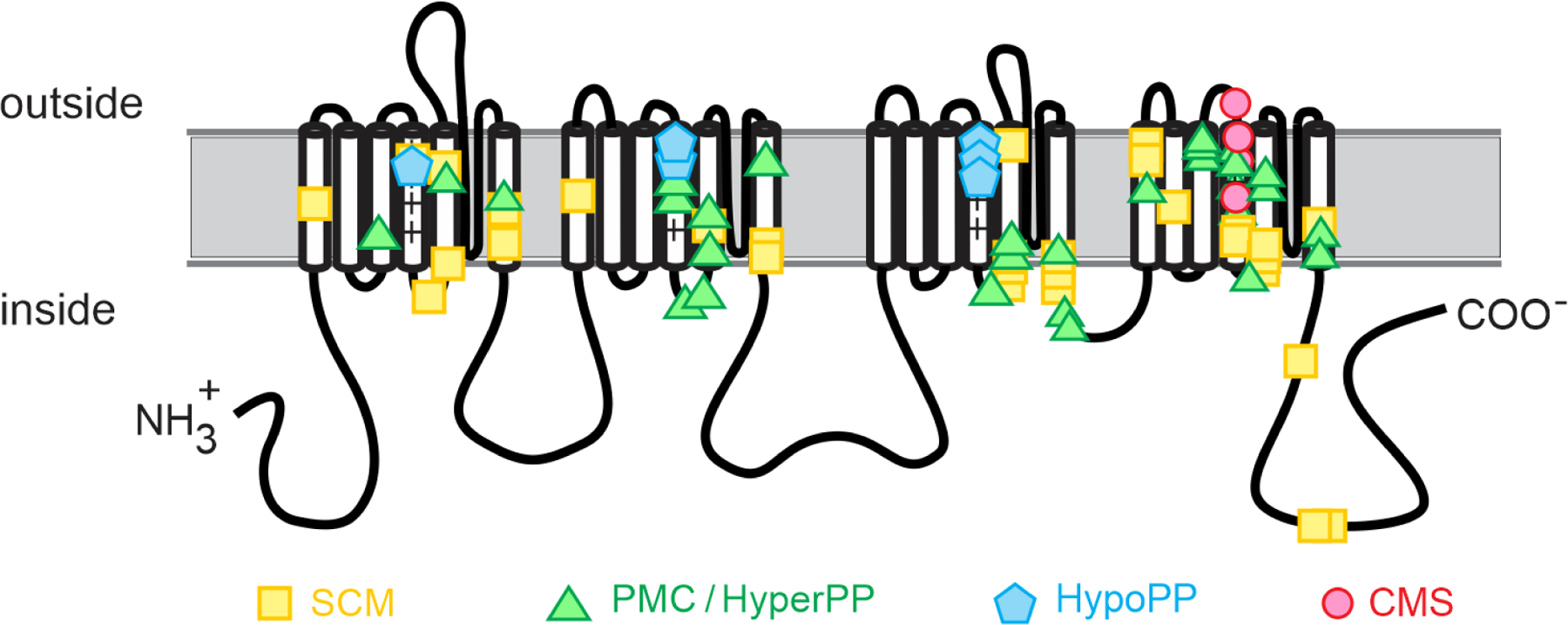
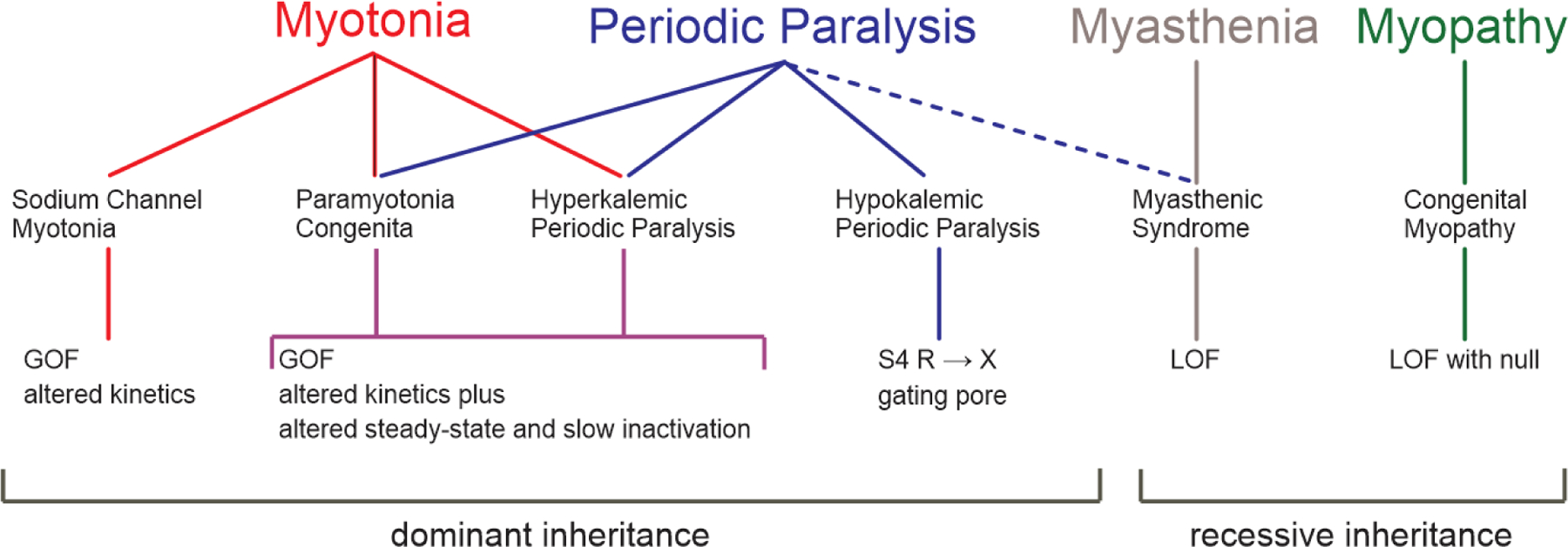
Periodic paralysis is a rare, dominantly inherited disorder of skeletal muscle in which episodic attacks of weakness are caused by a transient impairment of fiber excitability. Attacks of weakness are often elicited by characteristic environmental triggers, which were the basis for clinically delineating subtypes of periodic paralysis and are an important distinction for optimal disease management. All forms of familial periodic paralysis are caused by mutations of ion channels, often selectively expressed in skeletal muscle, that destabilize the resting potential. The missense mutations usually alter channel function through gain-of-function changes rather than producing a complete loss-of-function null. The knowledge of which channel gene harbors a variant, whether that variant is expected to (or known to) alter function, and how altered function impairs fiber excitability aides in the interpretation of patient signs and symptoms, the interpretation of gene test results, and how to optimize therapeutic intervention for symptom management and improve quality of life.
Keywords: CACNA1S; Calcium channel; Channelopathy; KCNJ2; Muscle; Myotonia; Potassium channel; SCN4A; Sodium channel.
Copyright © 2024 Elsevier B.V. All rights are reserved, including those for text and data mining, AI training, and similar technologies.
Figures
References
-
- Arimura K, Arimura Y, Ng AR, et al. (2007). Muscle membrane excitability after exercise in thyrotoxic periodic paralysis and thyrotoxicosis without periodic paralysis. Muscle & nerve 36: 784–788. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
