

Mutant huntingtin impairs neurodevelopment in human brain organoids through CHCHD2-mediated neurometabolic failure
- PMID: 39174523
- PMCID: PMC11341898
- DOI: 10.1038/s41467-024-51216-w
Mutant huntingtin impairs neurodevelopment in human brain organoids through CHCHD2-mediated neurometabolic failure
Abstract
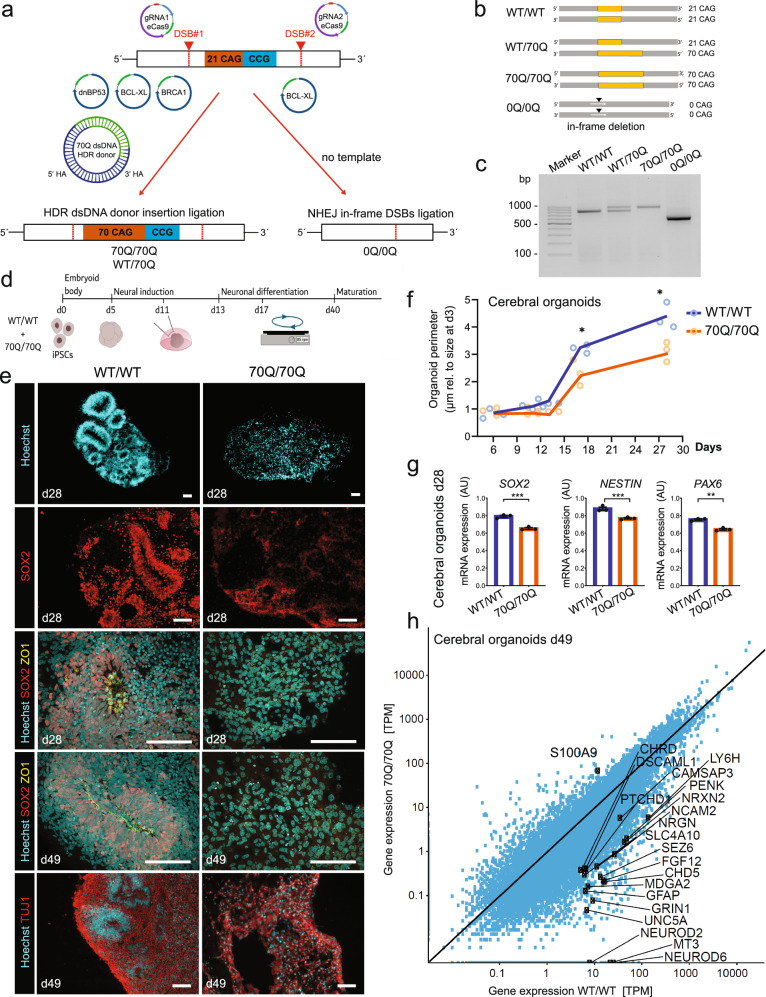
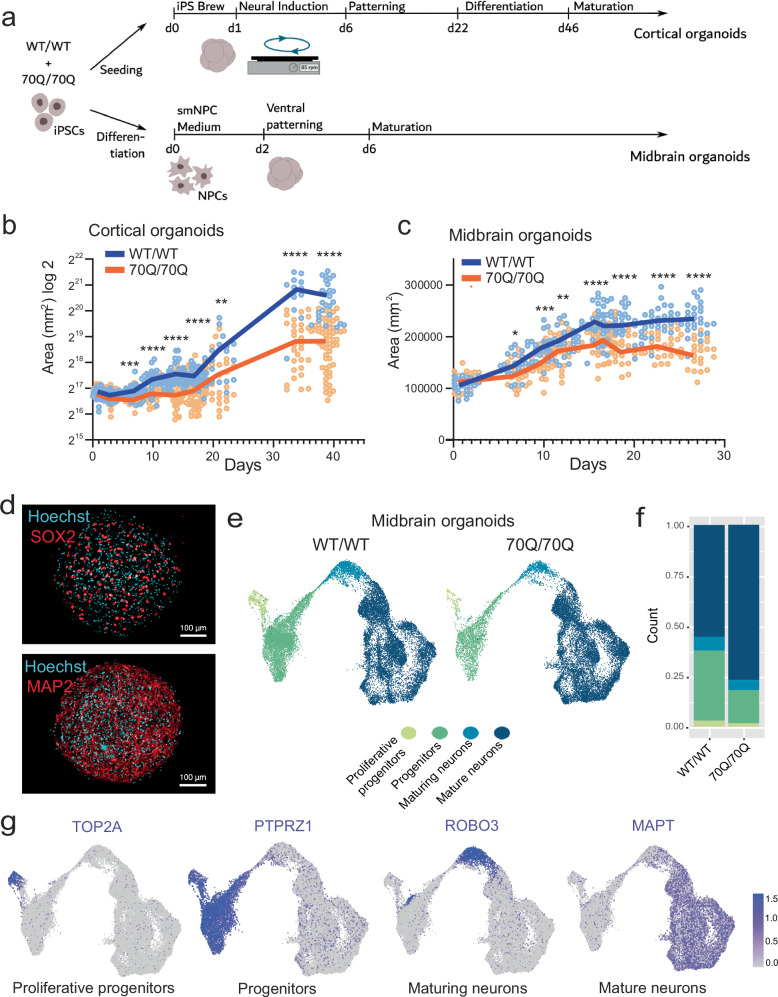
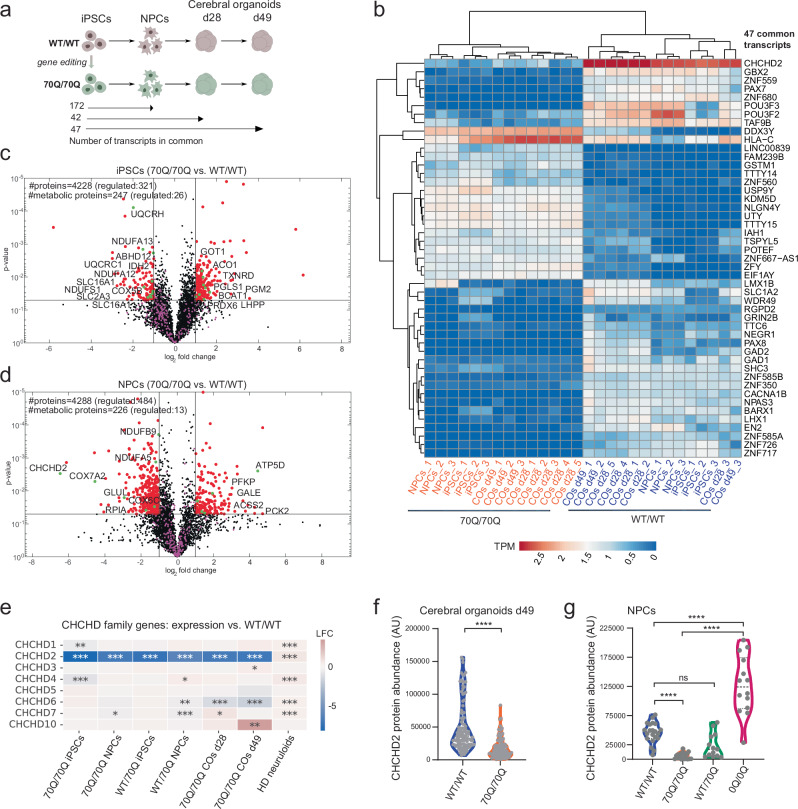
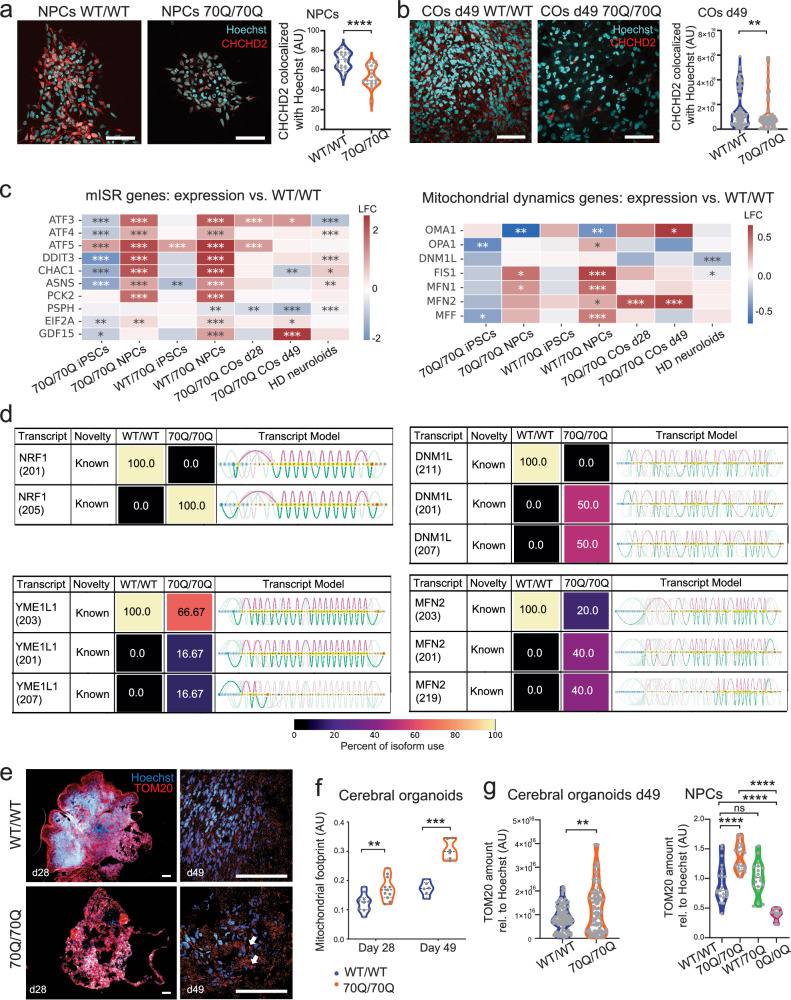
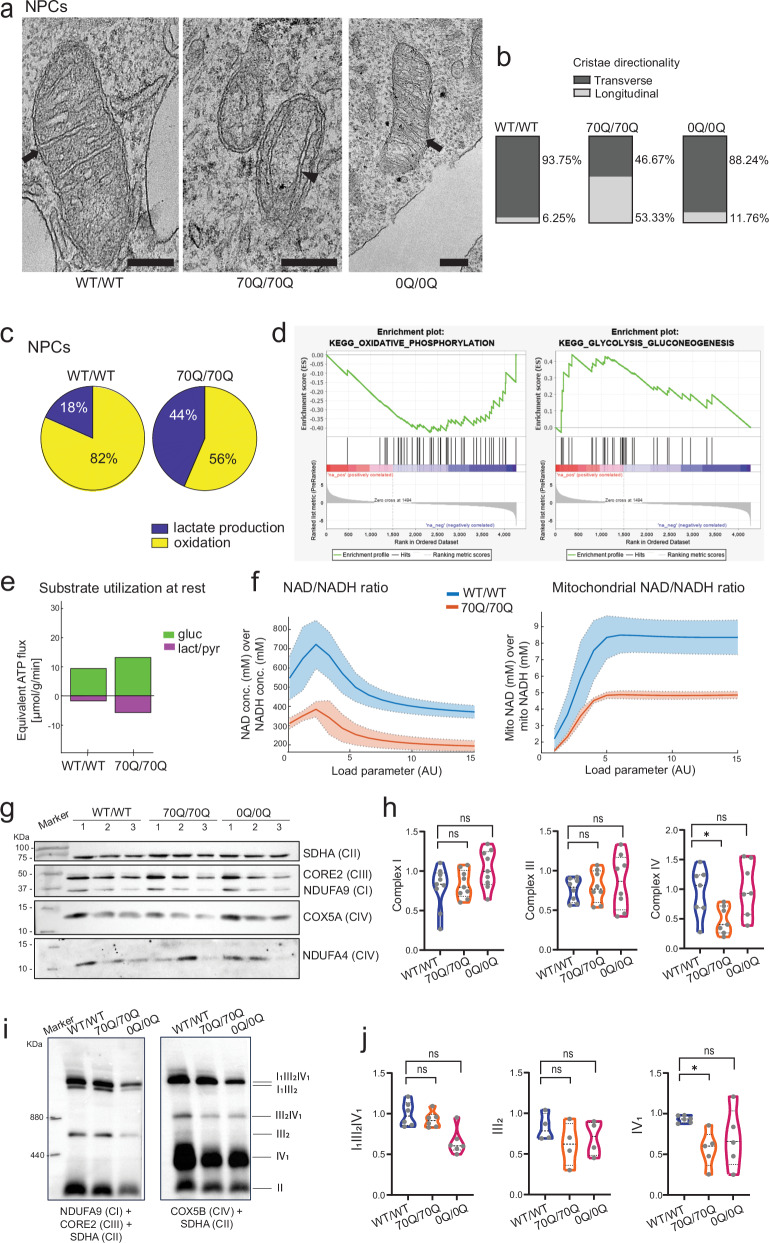
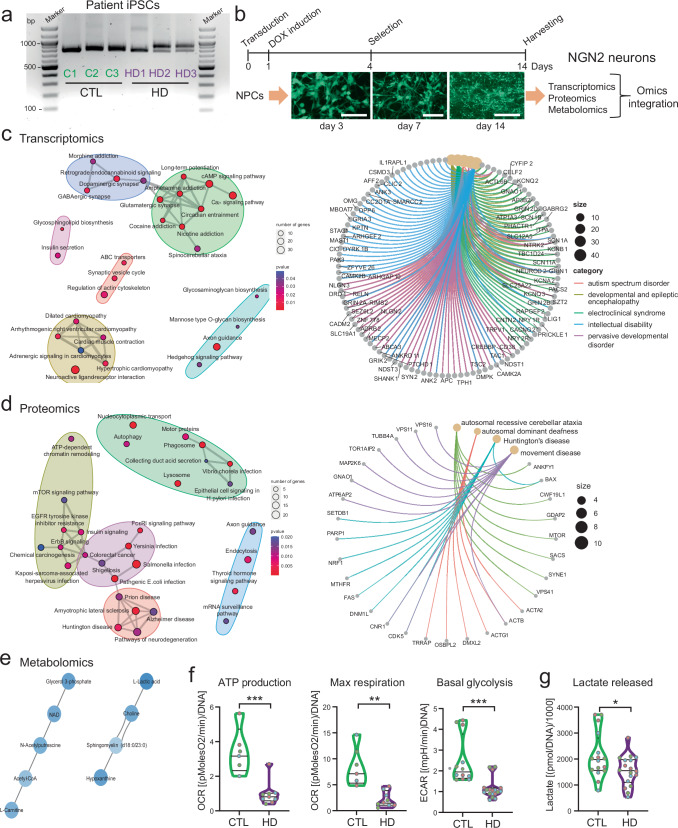
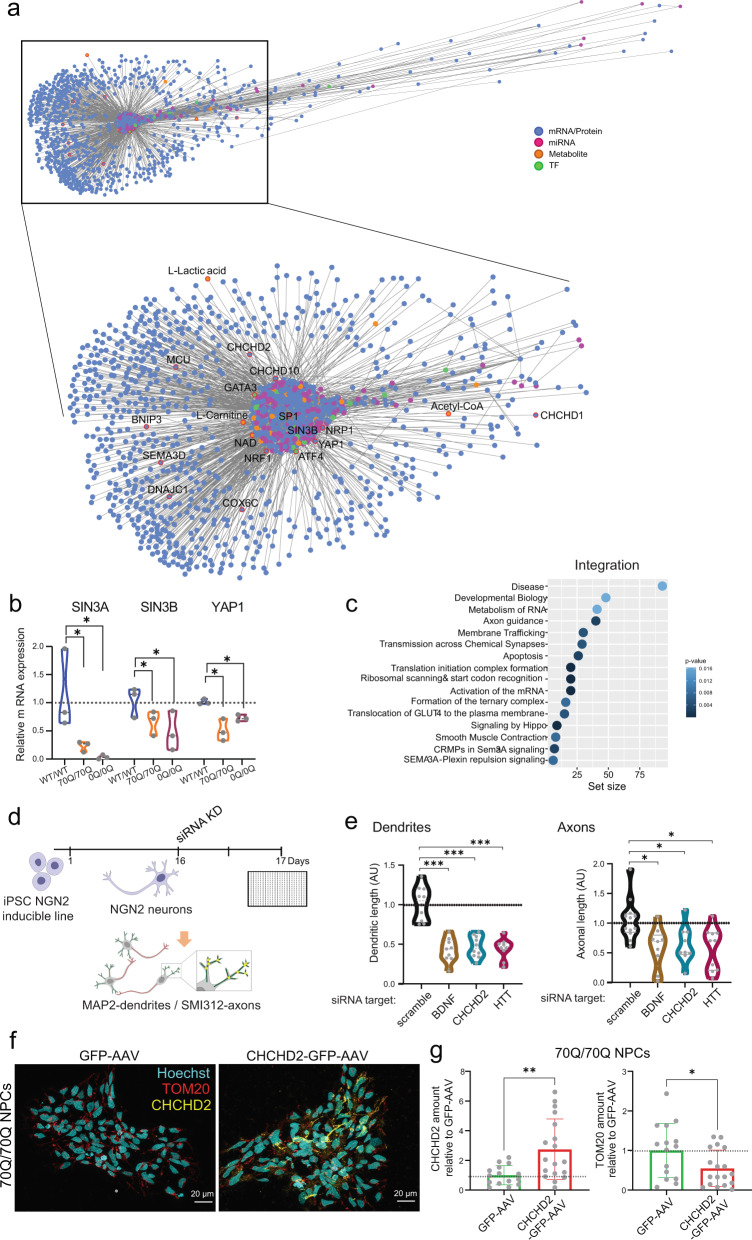
Expansion of the glutamine tract (poly-Q) in the protein huntingtin (HTT) causes the neurodegenerative disorder Huntington's disease (HD). Emerging evidence suggests that mutant HTT (mHTT) disrupts brain development. To gain mechanistic insights into the neurodevelopmental impact of human mHTT, we engineered male induced pluripotent stem cells to introduce a biallelic or monoallelic mutant 70Q expansion or to remove the poly-Q tract of HTT. The introduction of a 70Q mutation caused aberrant development of cerebral organoids with loss of neural progenitor organization. The early neurodevelopmental signature of mHTT highlighted the dysregulation of the protein coiled-coil-helix-coiled-coil-helix domain containing 2 (CHCHD2), a transcription factor involved in mitochondrial integrated stress response. CHCHD2 repression was associated with abnormal mitochondrial morpho-dynamics that was reverted upon overexpression of CHCHD2. Removing the poly-Q tract from HTT normalized CHCHD2 levels and corrected key mitochondrial defects. Hence, mHTT-mediated disruption of human neurodevelopment is paralleled by aberrant neurometabolic programming mediated by dysregulation of CHCHD2, which could then serve as an early interventional target for HD.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
- PR1527/5-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 01GM2002A/Bundesministerium für Bildung und Forschung (Federal Ministry of Education and Research)
- 101080249/EC | EU Framework Programme for Research and Innovation H2020 | H2020 European Institute of Innovation and Technology (H2020 The European Institute of Innovation and Technology)
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
