

Angelman syndrome in Poland: current diagnosis and therapy status-the caregiver perspective: a questionnaire study
- PMID: 39174987
- PMCID: PMC11340045
- DOI: 10.1186/s13023-024-03292-w
Angelman syndrome in Poland: current diagnosis and therapy status-the caregiver perspective: a questionnaire study
Abstract
Background: Angelman syndrome (AS) is a rare neurodevelopmental disease caused by imprinting disorders that impede the production of the ubiquitin E3A ligase protein (UBE3A). AS affects multiple systems, with the main symptoms including epilepsy, psychomotor disorders and speech development disorders. To date, no study has been conducted in the Polish population to verify the condition's diagnosis and treatment process.
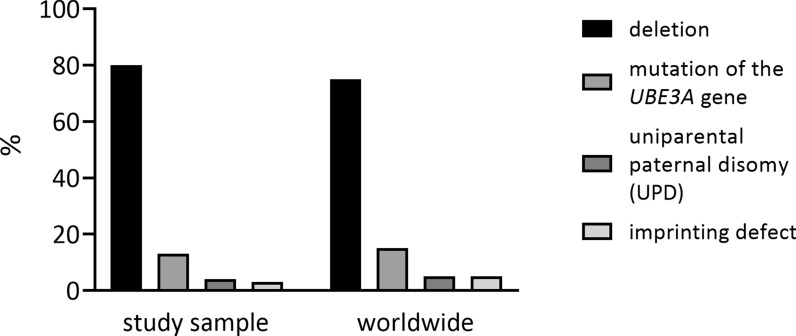
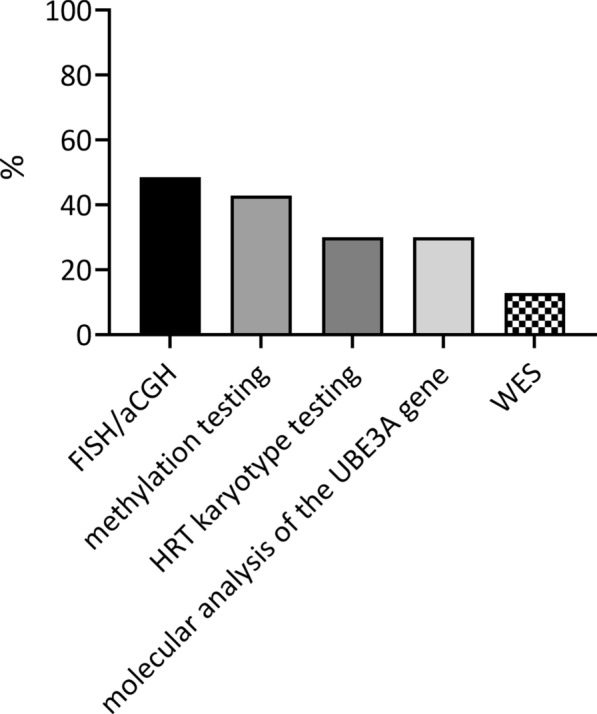
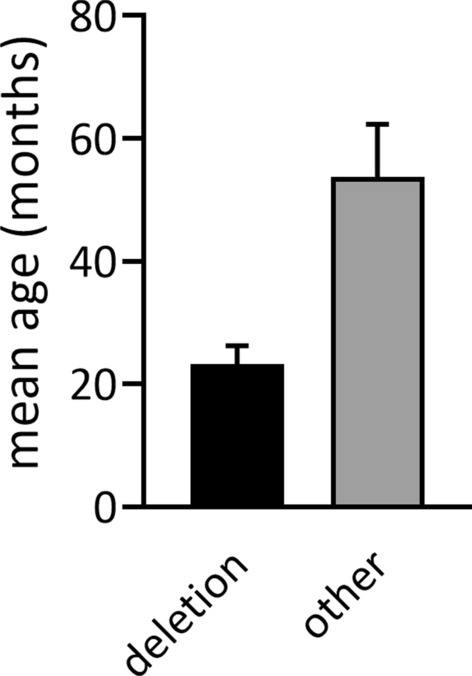
Results: Seventy patients with the median age of 60 months were included into the analysis. 80% of patients were diagnosed with deletion, 19.9% with a mutation of UBE3A gene, 4.3% with paternal uniparental disomy (UPD) and 2.8% with an imprinting defect. The mean age of first symptoms was 5 months, while the mean age of diagnosis was 29 months (earliest in deletion group at 23 months), and the median duration of diagnosis process was 7 months. The average time to a clinical geneticist appointment was 3 months. 37.9% of the patients initially received a different diagnosis. Epileptic seizures were present in 88.6% of the individuals. 98.6% of the studied group were under care of a pediatric neurologist, 47.1% of a gastroenterologist. A ketogenic diet was used in 7.1% of patients. Caregivers identified finding a specialist suitable for AS patients and access to genetic testing as the biggest problems.
Conclusions: The care of patients with AS in Poland is carried out according to the European and world standards, however there is an impeded access to clinical geneticist, and the knowledge about rare diseases among primary healthcare physicians could be improved. Moreover, access to AS care specialists and coordination of care is limited. There is a need for creation a specialized centers and databases for AS patients.
Keywords: Angelman syndrome; Genetic testing; Healthcare organisation; Rase diseases.
© 2024. The Author(s).
Conflict of interest statement
DC and KŁ are board members in PROT sp. z o.o. and Neurare Research Foundation.
Figures
References
-
- Obersztyn E. Angelman syndrome. In: Śmigiel R, Szczałuba K, editors. Genetically conditioned developmental disorders in children. I. Warsaw: PZWL; 2021. p. 958–76.
-
- Śmigiel R, Łukasiewicz K, Suleja A, et al. Diagnostic and therapeutic recommendations in Angelman syndrome. Pediatr Clin. 2023;31(Neurometabolism 2023):6085–92.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
