

Renal tubular epithelial cells response to injury in acute kidney injury
- PMID: 39178744
- PMCID: PMC11388183
- DOI: 10.1016/j.ebiom.2024.105294
Renal tubular epithelial cells response to injury in acute kidney injury
Abstract
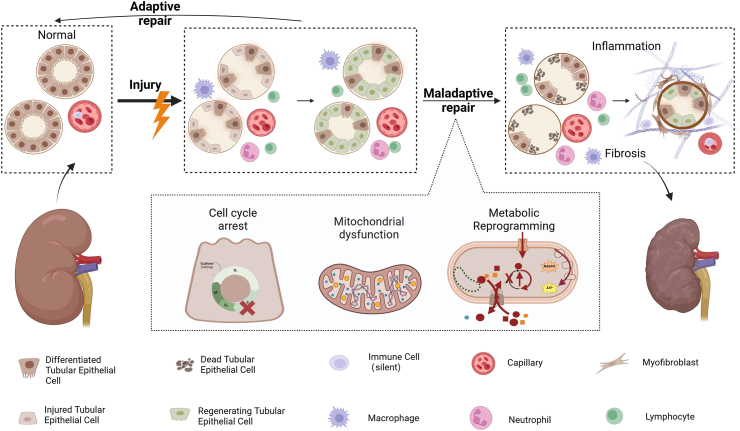
Acute kidney injury (AKI) is a clinical syndrome characterized by a rapid and significant decrease in renal function that can arise from various etiologies, and is associated with high morbidity and mortality. The renal tubular epithelial cells (TECs) represent the central cell type affected by AKI, and their notable regenerative capacity is critical for the recovery of renal function in afflicted patients. The adaptive repair process initiated by surviving TECs following mild AKI facilitates full renal recovery. Conversely, when injury is severe or persistent, it allows the TECs to undergo pathological responses, abnormal adaptive repair and phenotypic transformation, which will lead to the development of renal fibrosis. Given the implications of TECs fate after injury in renal outcomes, a deeper understanding of these mechanisms is necessary to identify promising therapeutic targets and biomarkers of the repair process in the human kidney.
Keywords: Acute kidney injury; Adaptive repair; Phenotypic transformation; Renal tubular epithelial cells.
Copyright © 2024 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests The authors confirm that there are no conflicts of interest.
Figures



References
-
- Matsuura R., Doi K., Rabb H. Acute kidney injury and distant organ dysfunction-network system analysis. Kidney Int. 2023;103(6):1041–1055. - PubMed
-
- Ronco C., Bellomo R., Kellum J.A. Acute kidney injury. Lancet. 2019;394(10212):1949–1964. - PubMed
-
- Li Z.L., Huang M.M., Yu M.Y., et al. Mitochondrial fumarate promotes ischemia/reperfusion-induced tubular injury. Acta Physiol (Oxf) 2024;240(4) - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
