

Unbiased discovery of cancer pathways and therapeutics using Pathway Ensemble Tool and Benchmark
- PMID: 39179644
- PMCID: PMC11343859
- DOI: 10.1038/s41467-024-51859-9
Unbiased discovery of cancer pathways and therapeutics using Pathway Ensemble Tool and Benchmark
Abstract
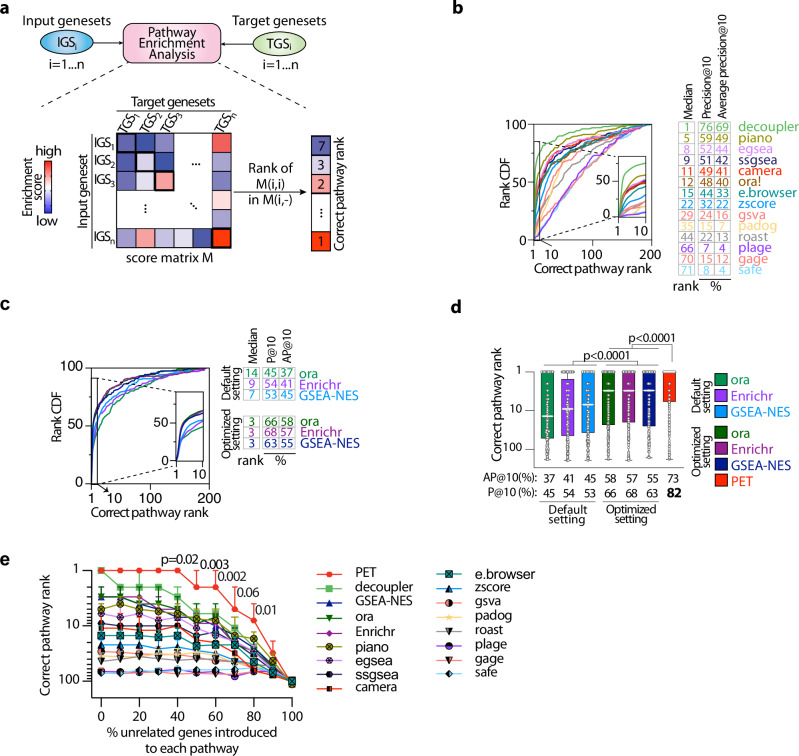
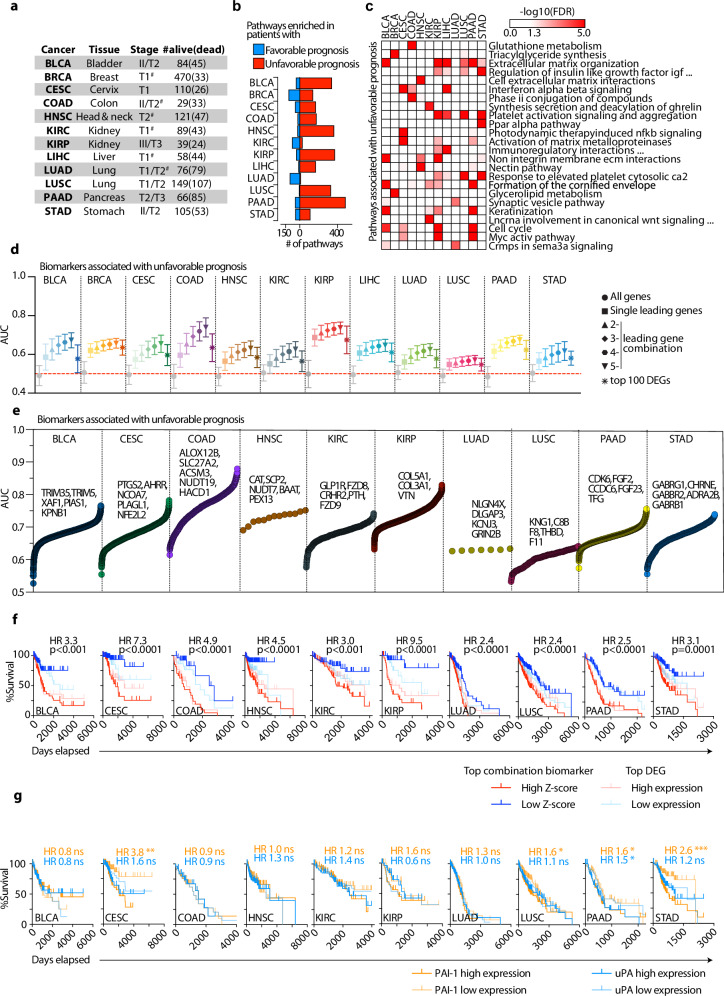
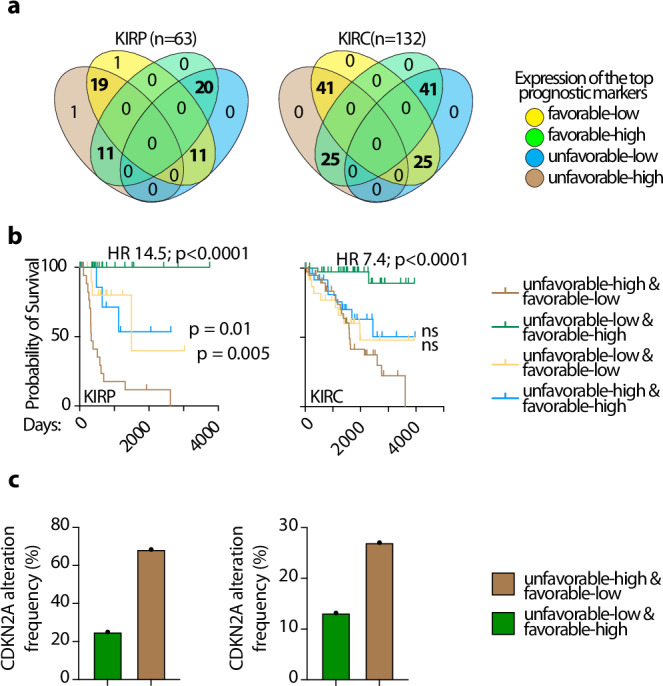
Correctly identifying perturbed biological pathways is a critical step in uncovering basic disease mechanisms and developing much-needed therapeutic strategies. However, whether current tools are optimal for unbiased discovery of relevant pathways remains unclear. Here, we create "Benchmark" to critically evaluate existing tools and find that most function sub-optimally. We thus develop the "Pathway Ensemble Tool" (PET), which outperforms existing methods. Deploying PET, we identify prognostic pathways across 12 cancer types. PET-identified prognostic pathways offer additional insights, with genes within these pathways serving as reliable biomarkers for clinical outcomes. Additionally, normalizing these pathways using drug repurposing strategies represents therapeutic opportunities. For example, the top predicted repurposed drug for bladder cancer, a CDK2/9 inhibitor, represses cell growth in vitro and in vivo. We anticipate that using Benchmark and PET for unbiased pathway discovery will offer additional insights into disease mechanisms across a spectrum of diseases, enabling biomarker discovery and therapeutic strategies.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Howlader, N. et al. SEER Cancer Statistics Review, 1975–2018. National Cancer Institute. Bethesda, MD, https://seer.cancer.gov/csr/1975_2018/, based on November 2020 SEER data submission posted to the SEER web site (2021).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
