

Mapping spatial organization and genetic cell-state regulators to target immune evasion in ovarian cancer
- PMID: 39179931
- PMCID: PMC11436371
- DOI: 10.1038/s41590-024-01943-5
Mapping spatial organization and genetic cell-state regulators to target immune evasion in ovarian cancer
Abstract
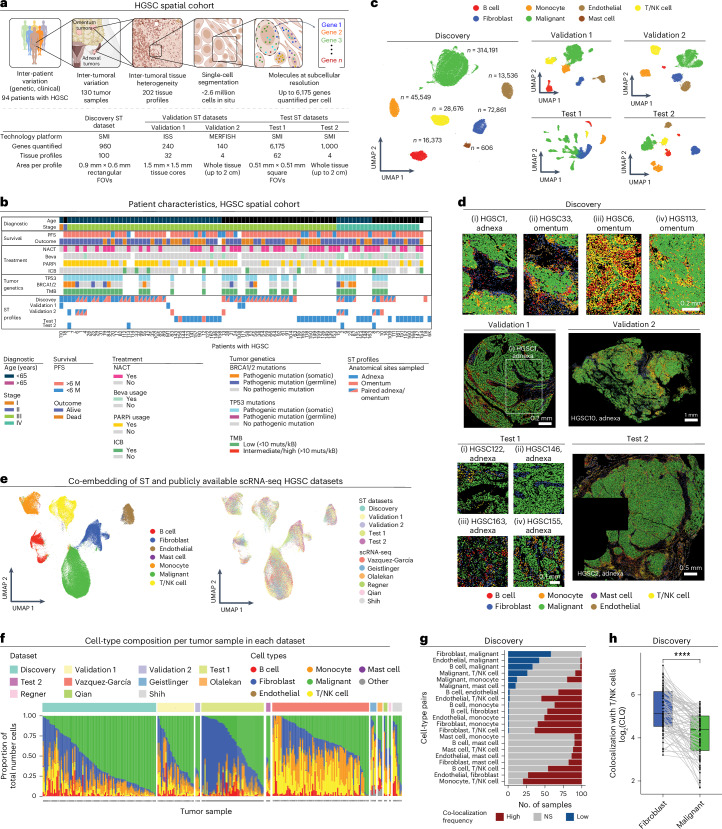
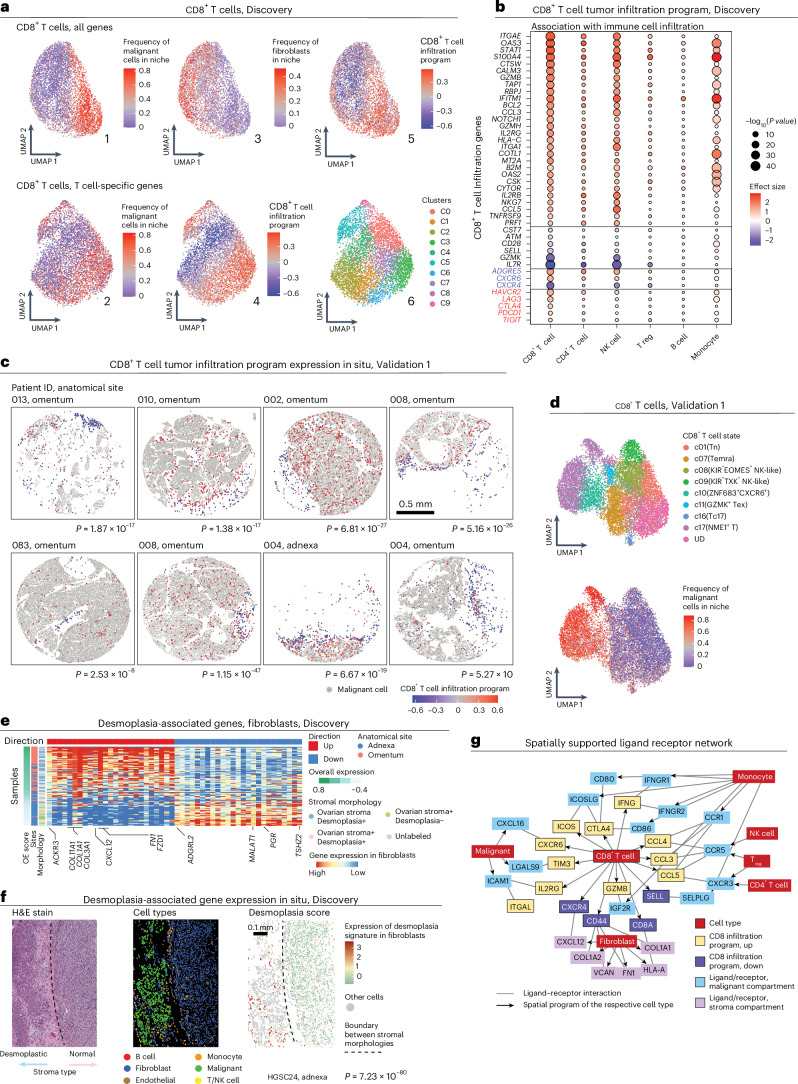
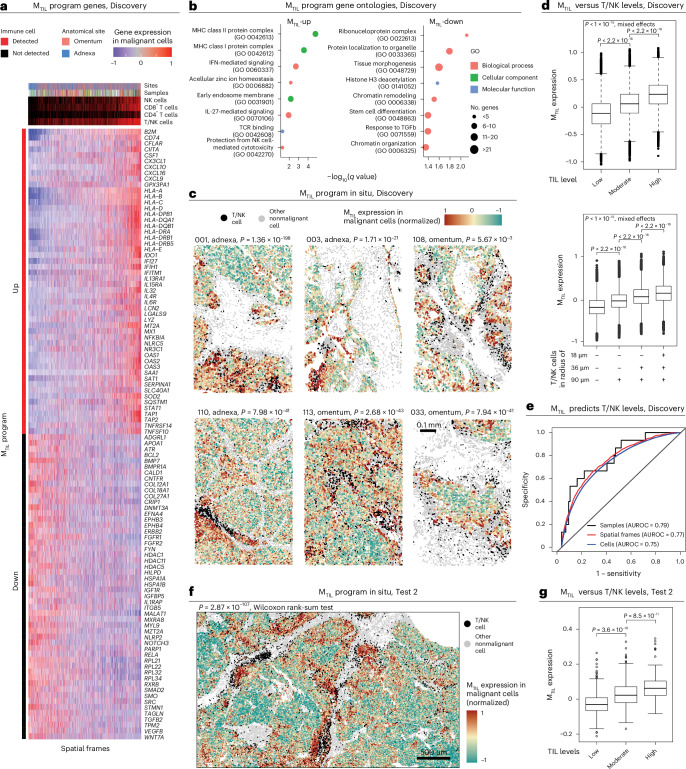
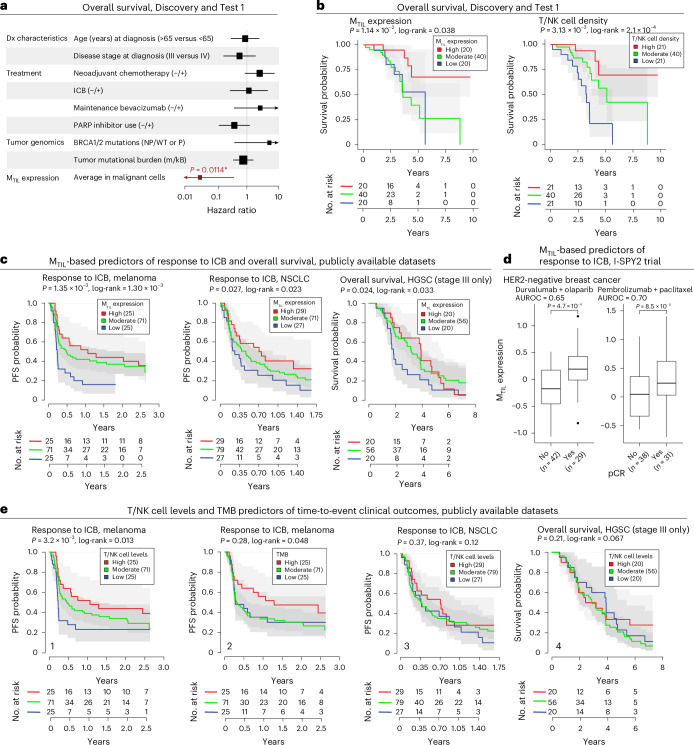
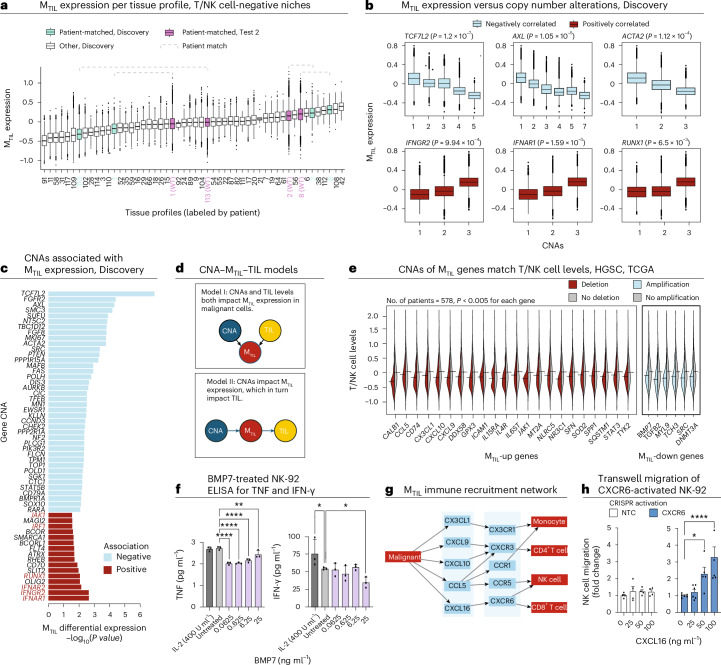
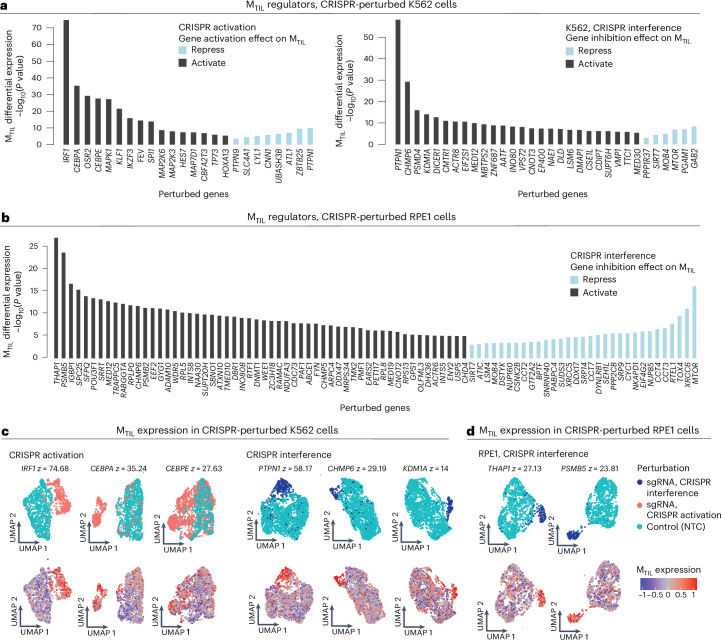
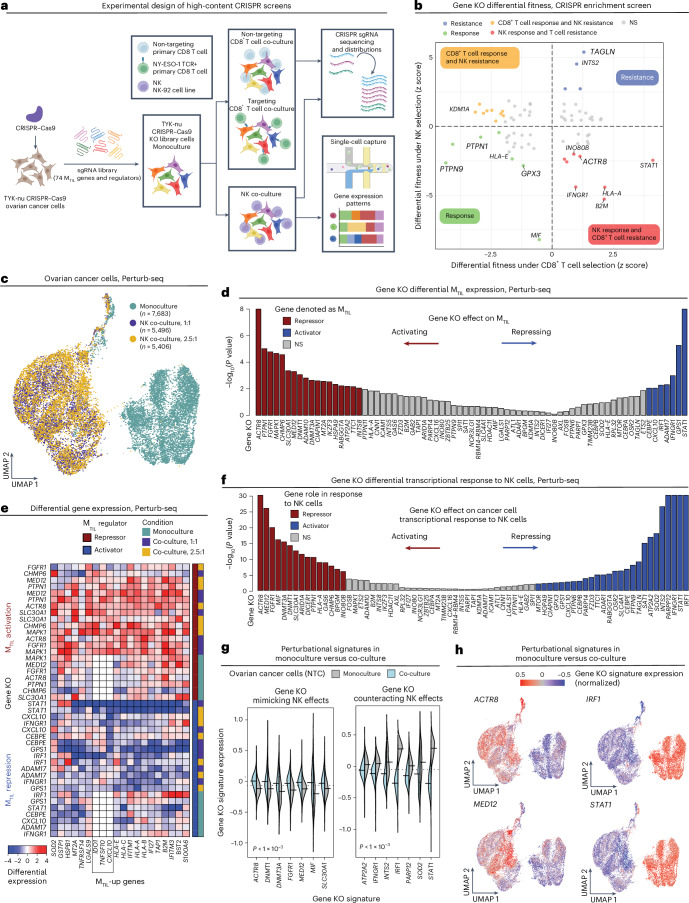
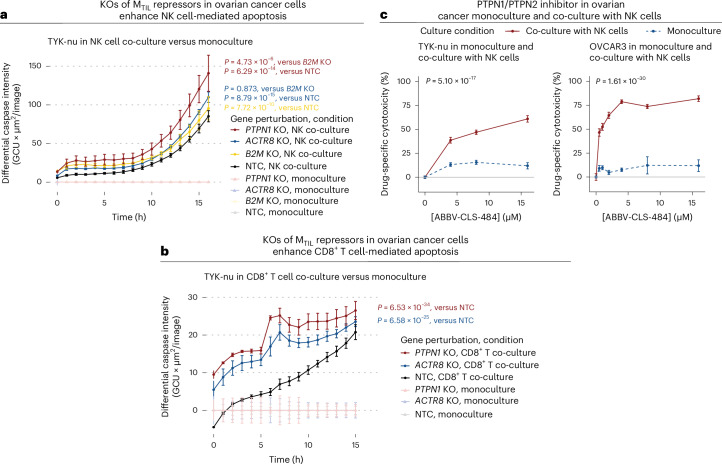
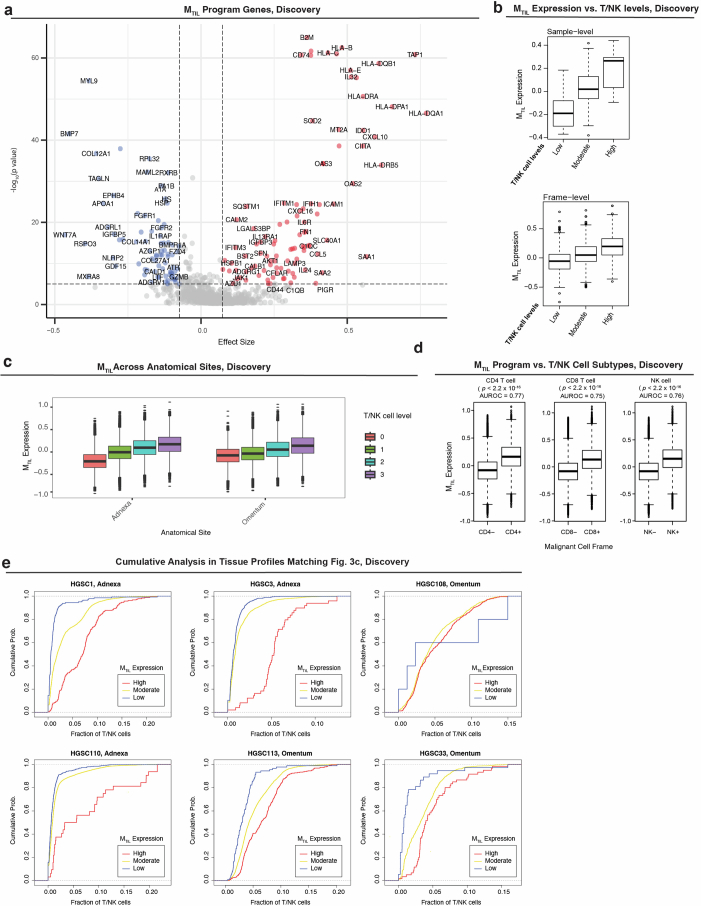
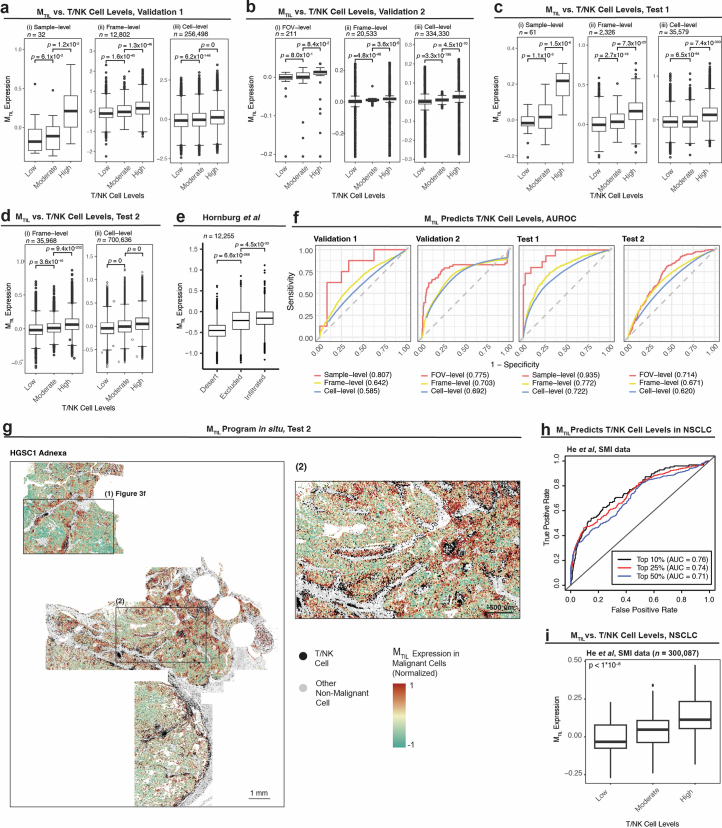
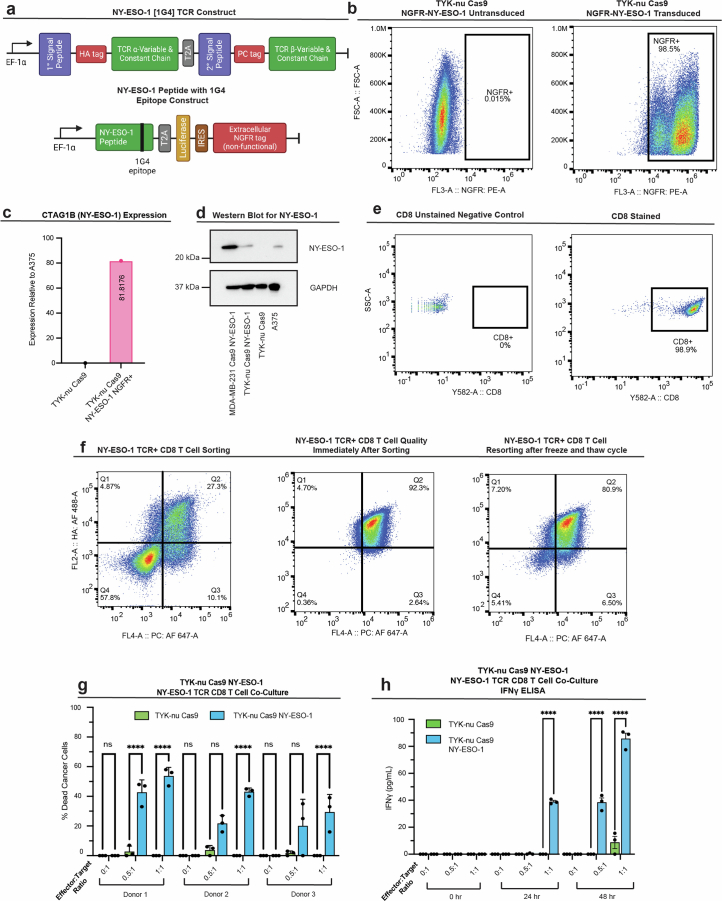
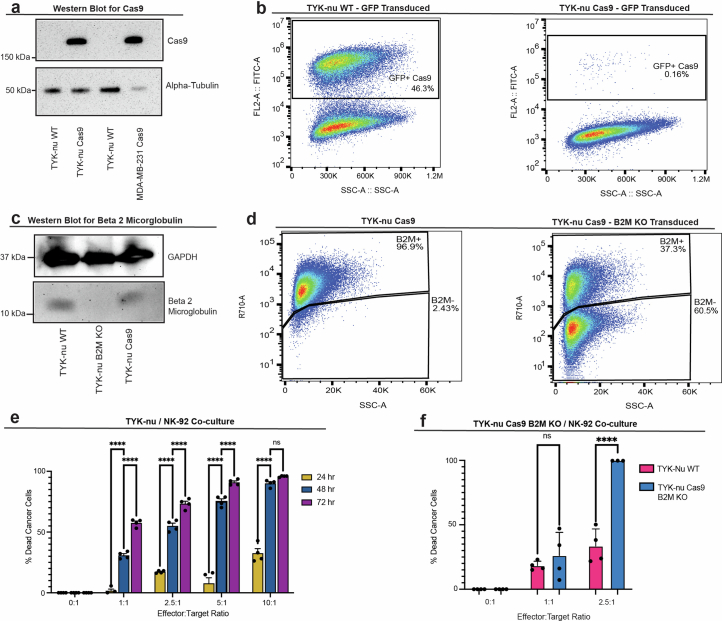
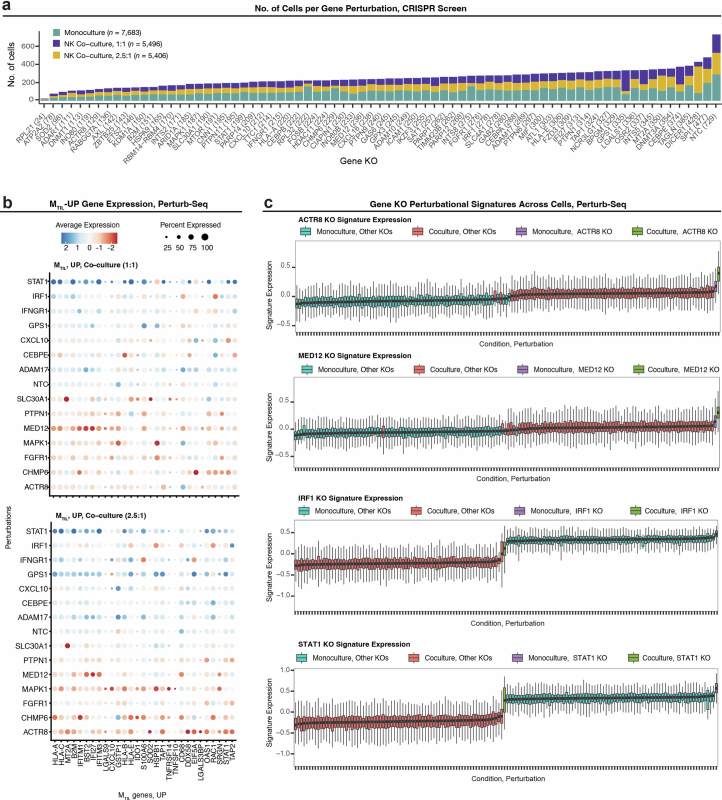
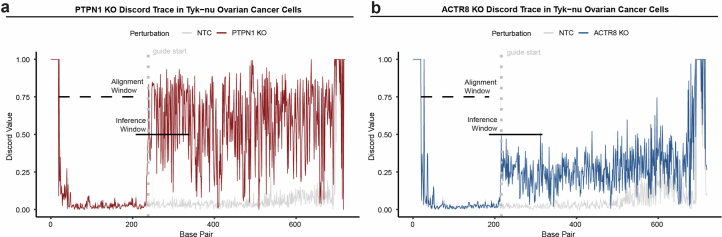
The drivers of immune evasion are not entirely clear, limiting the success of cancer immunotherapies. Here we applied single-cell spatial and perturbational transcriptomics to delineate immune evasion in high-grade serous tubo-ovarian cancer. To this end, we first mapped the spatial organization of high-grade serous tubo-ovarian cancer by profiling more than 2.5 million cells in situ in 130 tumors from 94 patients. This revealed a malignant cell state that reflects tumor genetics and is predictive of T cell and natural killer cell infiltration levels and response to immune checkpoint blockade. We then performed Perturb-seq screens and identified genetic perturbations-including knockout of PTPN1 and ACTR8-that trigger this malignant cell state. Finally, we show that these perturbations, as well as a PTPN1/PTPN2 inhibitor, sensitize ovarian cancer cells to T cell and natural killer cell cytotoxicity, as predicted. This study thus identifies ways to study and target immune evasion by linking genetic variation, cell-state regulators and spatial biology.
© 2024. The Author(s).
Conflict of interest statement
M.P.S. is a cofounder and scientific advisor of Personalis, SensOmics, Qbio, January AI, Fodsel, Filtricine, Protos, RTHM, Iollo, Marble Therapeutics and Mirvie. M.P.S. is a scientific advisor of Yuvan, Jupiter, Neuvivo, Swaza and Mitrix. M.C.B has outside interest in DEM Biopharma. The other authors declare no competing interests.
Figures
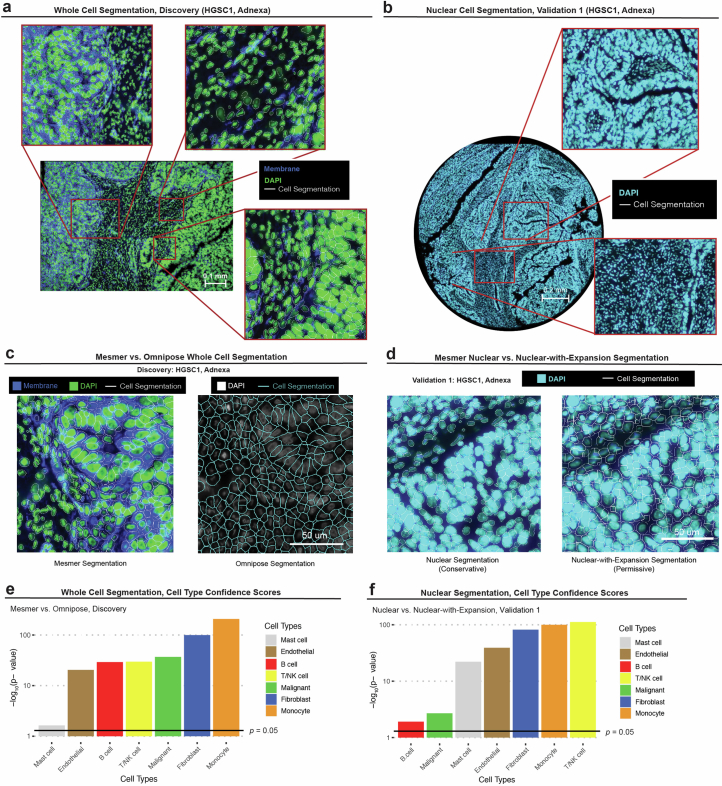
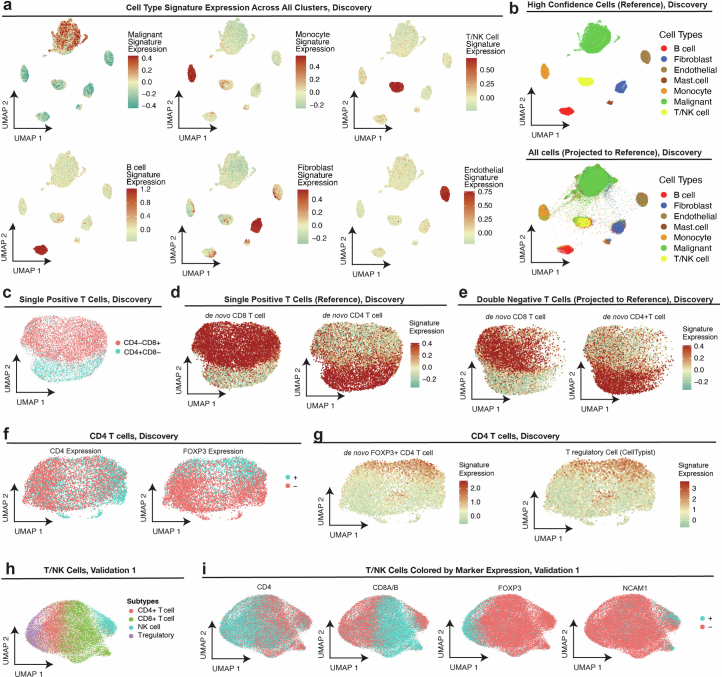
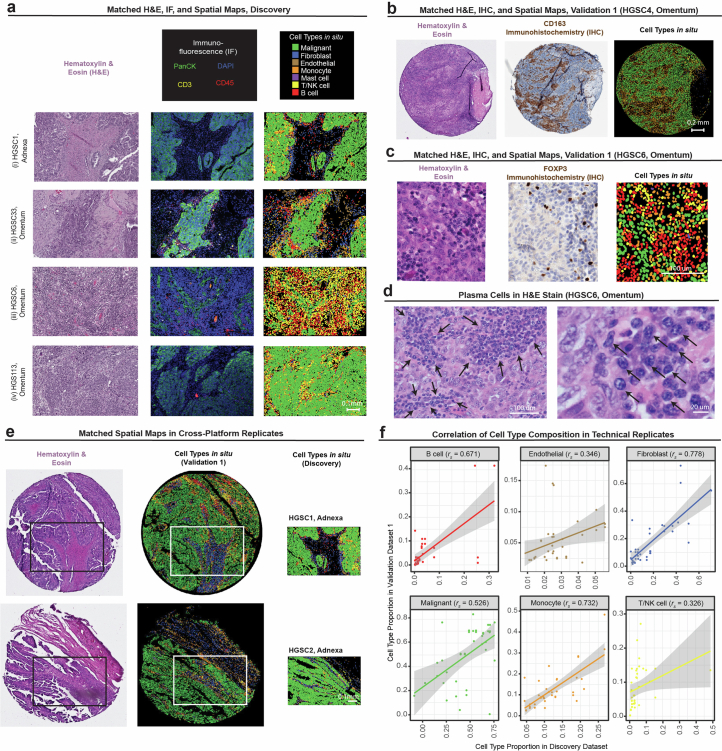
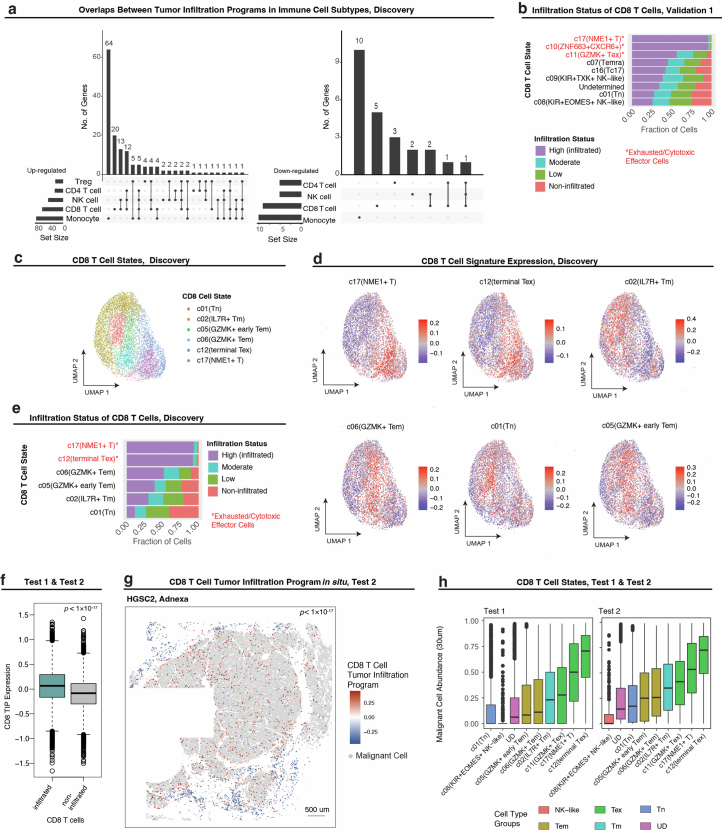
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
