

ΔNp63 bookmarks and creates an accessible epigenetic environment for TGFβ-induced cancer cell stemness and invasiveness
- PMID: 39180088
- PMCID: PMC11342681
- DOI: 10.1186/s12964-024-01794-5
ΔNp63 bookmarks and creates an accessible epigenetic environment for TGFβ-induced cancer cell stemness and invasiveness
Abstract
Background: p63 is a transcription factor with intrinsic pioneer factor activity and pleiotropic functions. Transforming growth factor β (TGFβ) signaling via activation and cooperative action of canonical, SMAD, and non-canonical, MAP-kinase (MAPK) pathways, elicits both anti- and pro-tumorigenic properties, including cell stemness and invasiveness. TGFβ activates the ΔNp63 transcriptional program in cancer cells; however, the link between TGFβ and p63 in unmasking the epigenetic landscape during tumor progression allowing chromatin accessibility and gene transcription, is not yet reported.
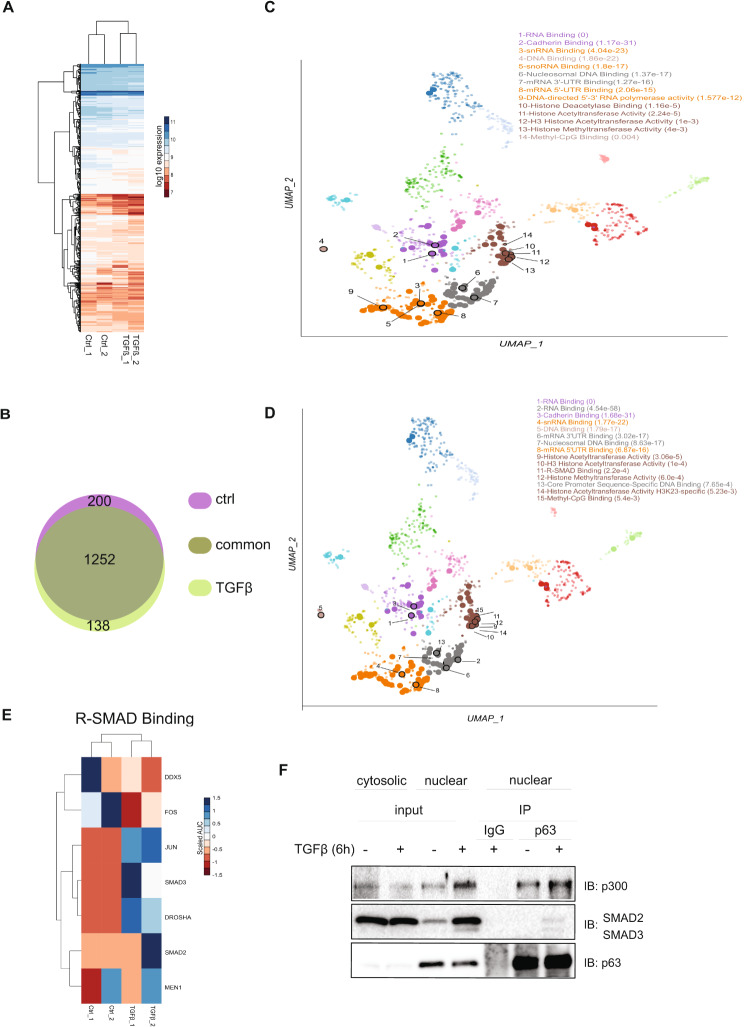
Methods: Small molecule inhibitors, including protein kinase inhibitors and RNA-silencing, provided loss of function analyses. Sphere formation assays in cancer cells, chromatin immunoprecipitation and mRNA expression assays were utilized in order to gain mechanistic evidence. Mass spectrometry analysis coupled to co-immunoprecipitation assays revealed novel p63 interactors and their involvement in p63-dependent transcription.
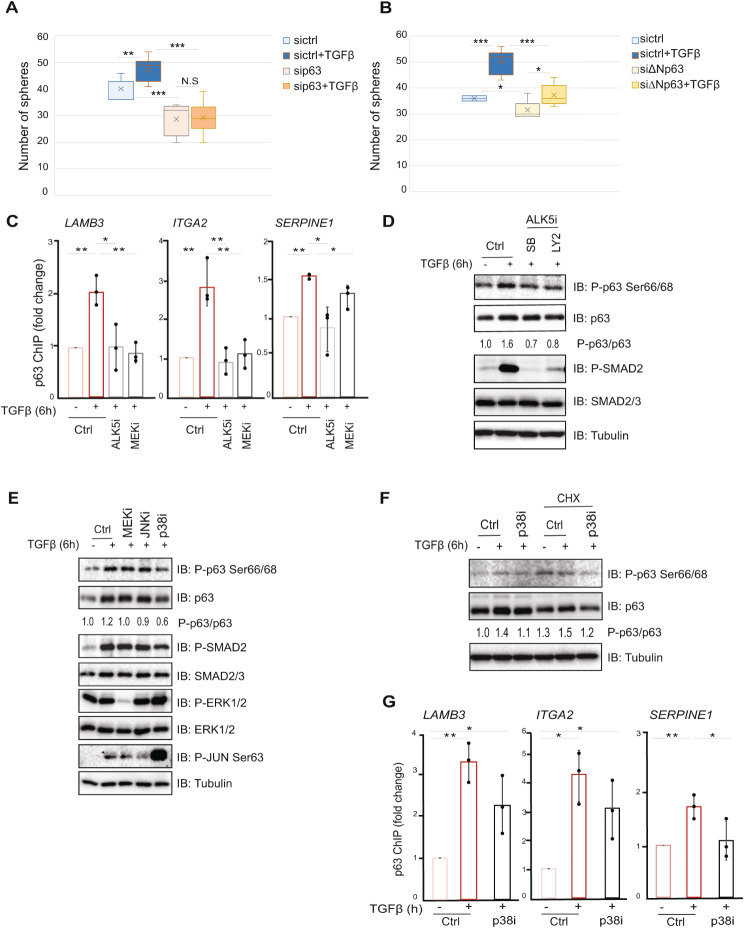
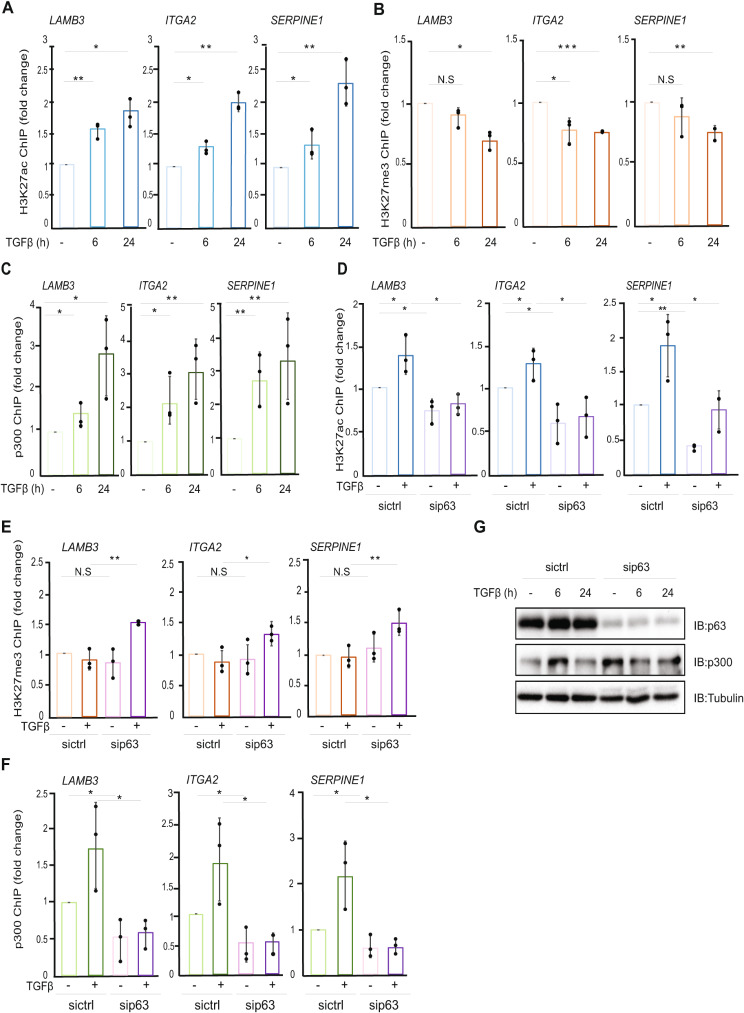
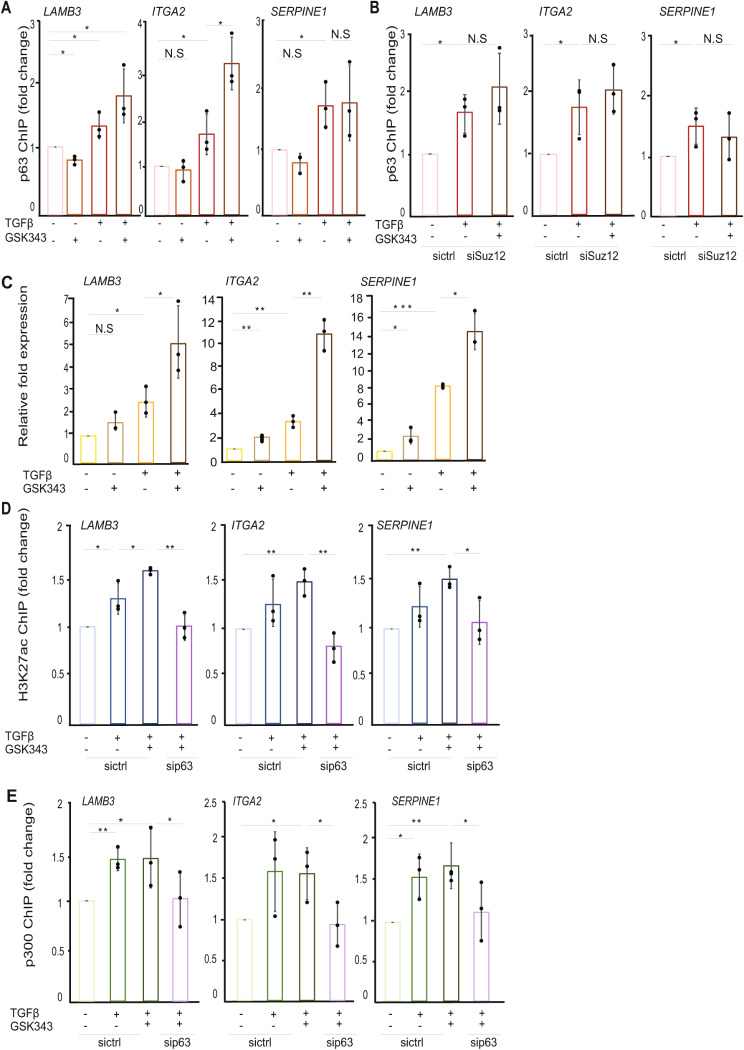
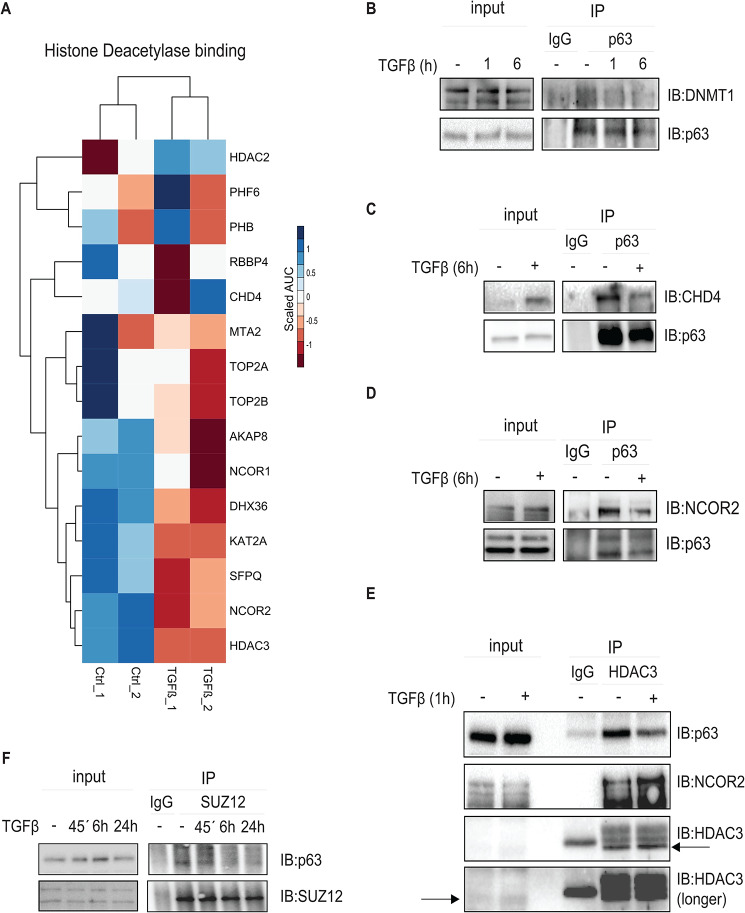
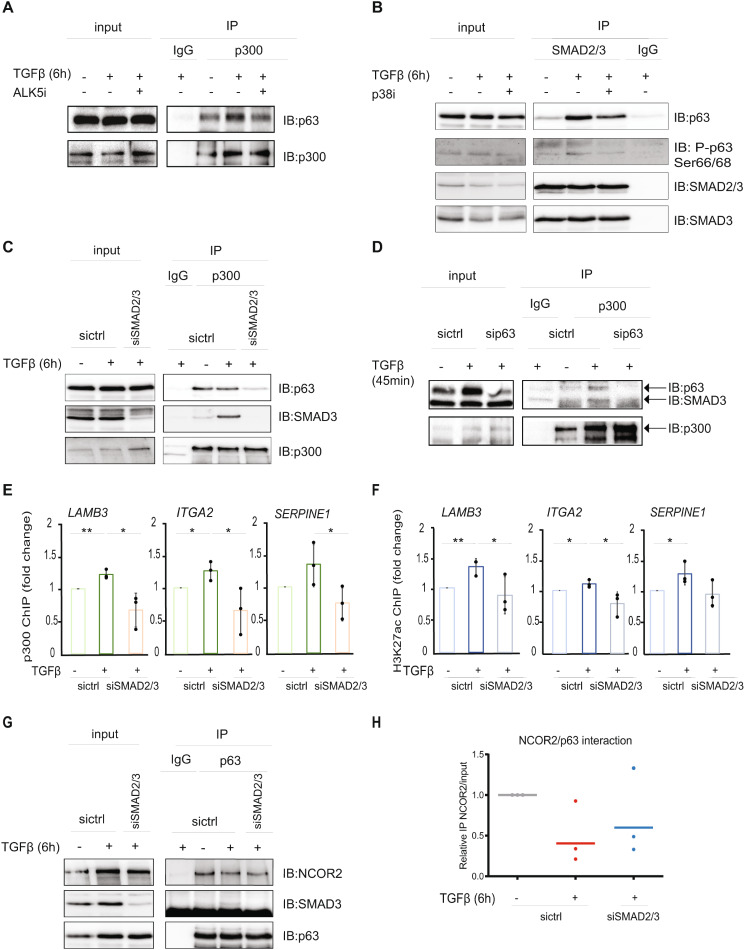
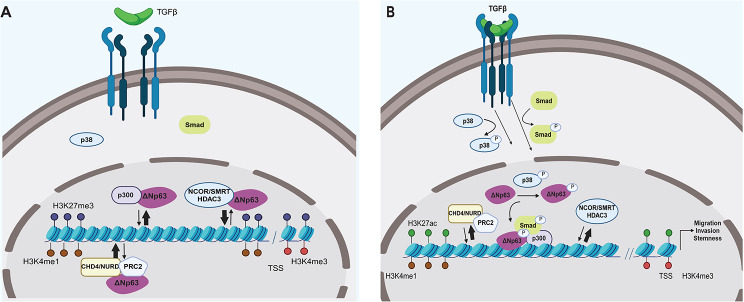
Results: The sphere-forming capacity of breast cancer cells was enhanced upon TGFβ stimulation and significantly decreased upon ΔNp63 depletion. Activation of TGFβ signaling via p38 MAPK signaling induced ΔNp63 phosphorylation at Ser 66/68 resulting in stabilized ΔNp63 protein with enhanced DNA binding properties. TGFβ stimulation altered the ratio of H3K27ac and H3K27me3 histone modification marks, pointing towards higher H3K27ac and increased p300 acetyltransferase recruitment to chromatin. By silencing the expression of ΔNp63, the TGFβ effect on chromatin remodeling was abrogated. Inhibition of H3K27me3, revealed the important role of TGFβ as the upstream signal for guiding ΔNp63 to the TGFβ/SMAD gene loci, as well as the indispensable role of ΔNp63 in recruiting histone modifying enzymes, such as p300, to these genomic regions, regulating chromatin accessibility and gene transcription. Mechanistically, TGFβ through SMAD activation induced dissociation of ΔNp63 from NURD or NCOR/SMRT histone deacetylation complexes, while promoted the assembly of ΔNp63-p300 complexes, affecting the levels of histone acetylation and the outcome of ΔNp63-dependent transcription.
Conclusions: ΔNp63, phosphorylated and recruited by TGFβ to the TGFβ/SMAD/ΔNp63 gene loci, promotes chromatin accessibility and transcription of target genes related to stemness and cell invasion.
Keywords: Chromatin accessibility; Protein-protein interaction; Signal transduction; Transcription; Transforming growth factor β (TGFβ); p63.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
