

Characterization of pathogenic genetic variants in Russian patients with primary ciliary dyskinesia using gene panel sequencing and transcript analysis
- PMID: 39180133
- PMCID: PMC11344339
- DOI: 10.1186/s13023-024-03318-3
Characterization of pathogenic genetic variants in Russian patients with primary ciliary dyskinesia using gene panel sequencing and transcript analysis
Abstract
Background: Primary ciliary dyskinesia (PCD) is a group of rare genetically heterogeneous disorders caused by defective cilia and flagella motility. The clinical phenotype of PCD patients commonly includes chronic oto-sino-pulmonary disease, infertility, and, in about half of cases, laterality defects due to randomization of left-right body asymmetry. To date, pathogenic variants in more than 50 genes responsible for motile cilia structure and assembly have been reported in such patients. While multiple population-specific mutations have been described in PCD cohorts from different countries, the data on genetic spectrum of PCD in Russian population are still extremely limited.
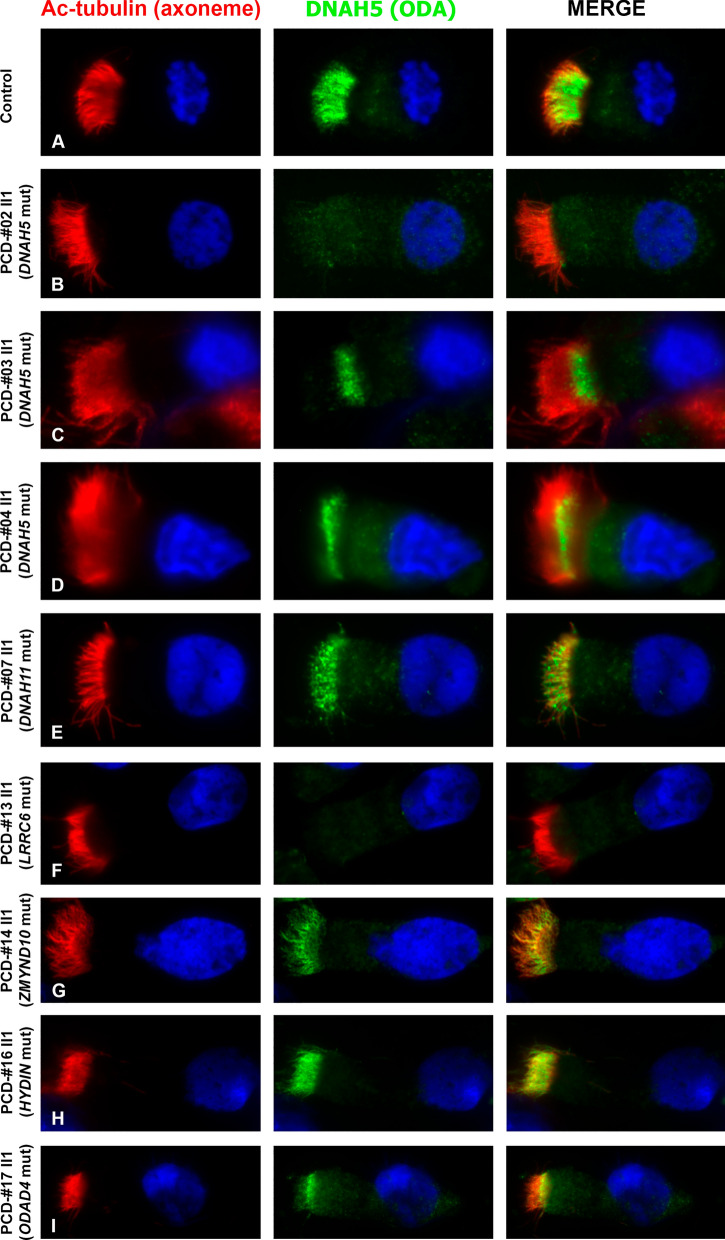
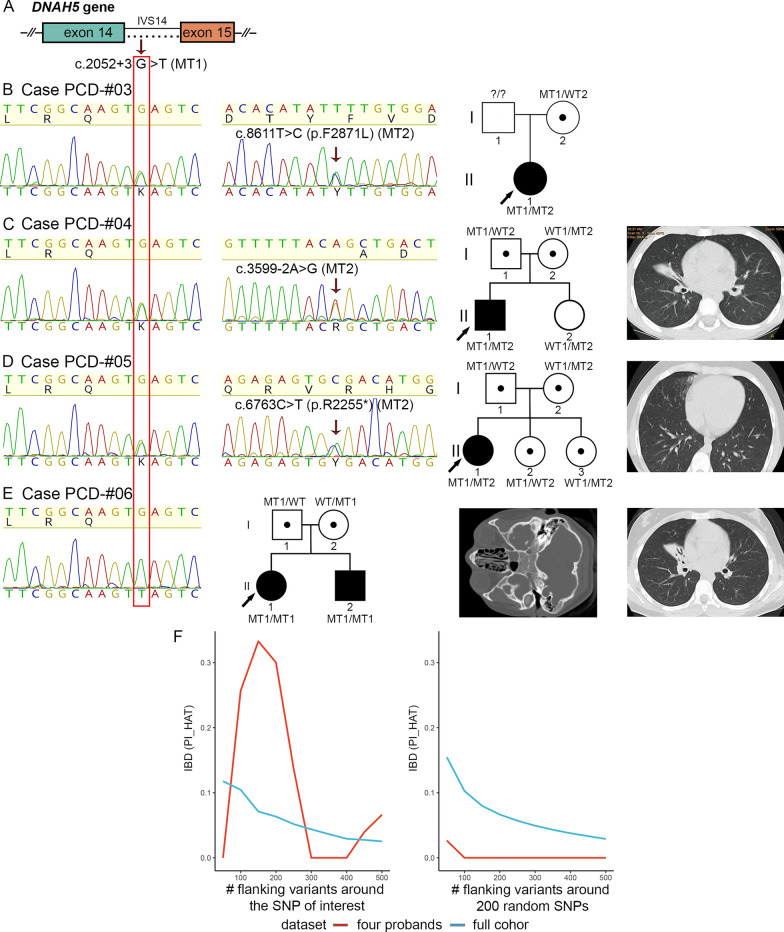
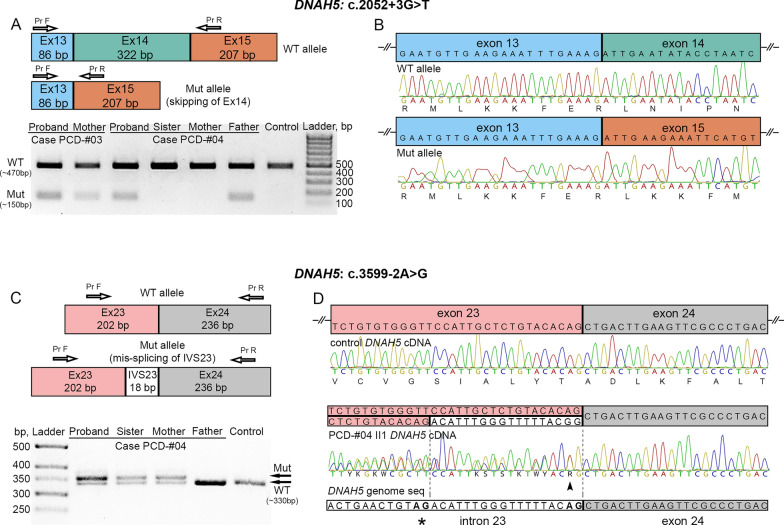
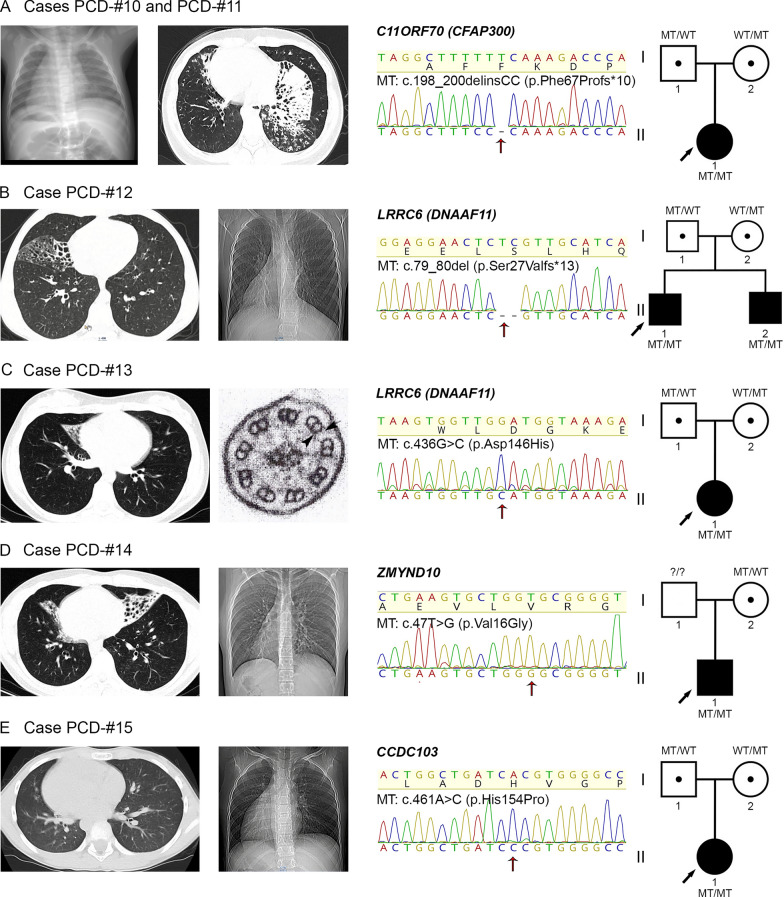
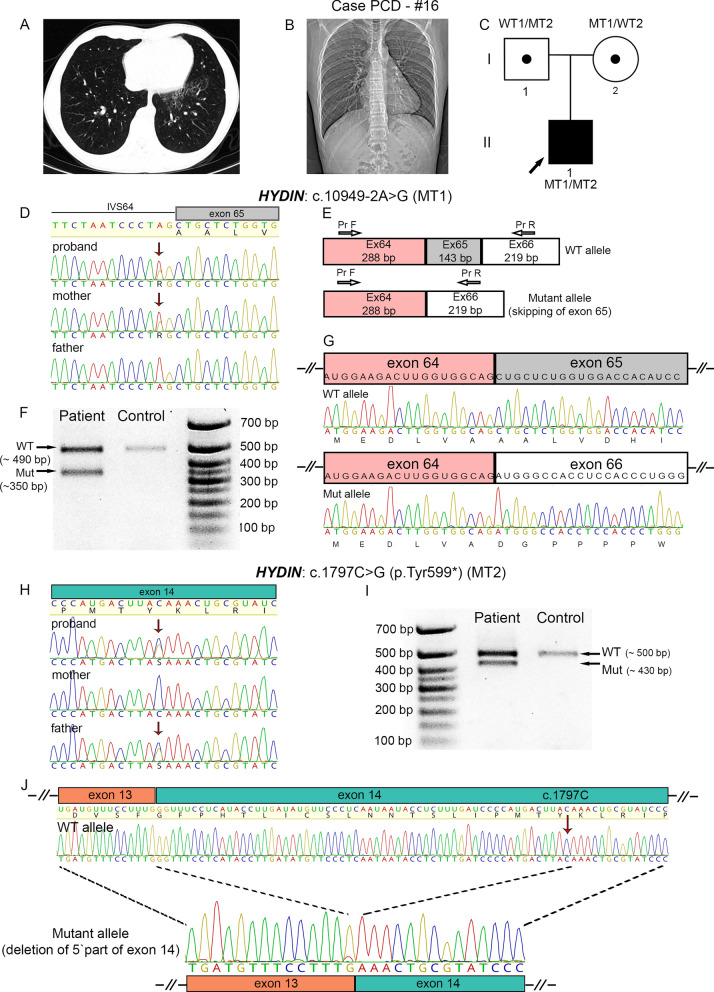
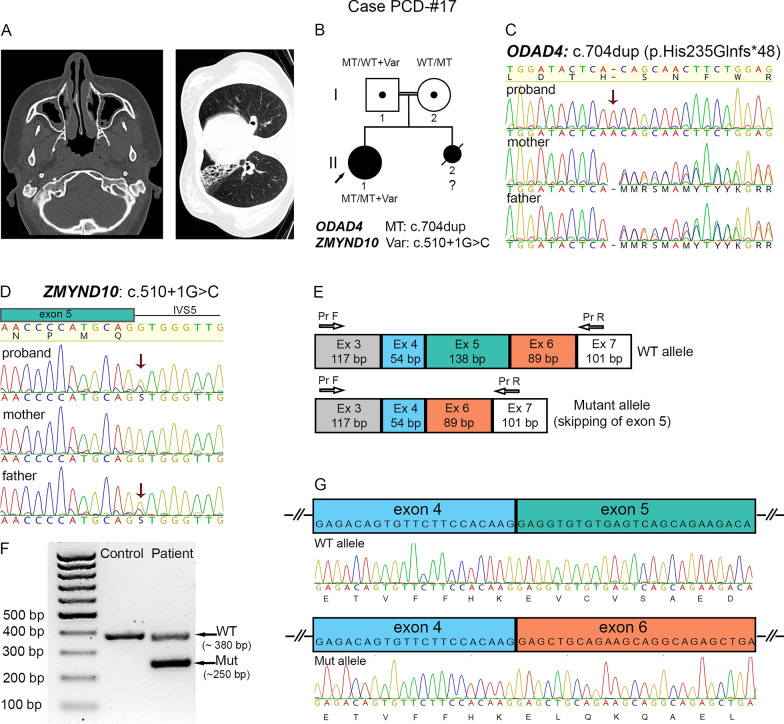
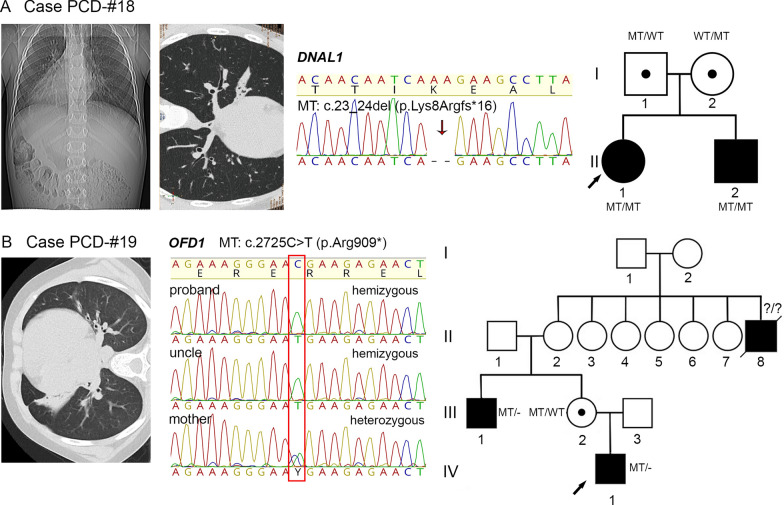
Results: The present study provides a comprehensive clinical and genetic characterization of 21 Russian families with PCD living in various country regions. Anomalies of ciliary beating in patients` respiratory epithelial cells were confirmed by high-speed video microscopy. In the most cases, custom-designed panel sequencing allowed to uncover causative variants in well-known or rarely mentioned PCD-related genes, including DNAH5, DNAH11, CFAP300, LRRC6, ZMYND10, CCDC103, HYDIN, ODAD4, DNAL1, and OFD1. The variations comprised common mutations, as well as novel genetic variants, some of which probably specific for Russian patients. Additional targeted analysis of mRNA transcripts from ciliated cells enabled us to specify functional effects of newly identified genetic variants in DNAH5 (c.2052+3G>T, c.3599-2A>G), HYDIN (c.10949-2A>G, c.1797C>G), and ZMYND10 (c.510+1G>C) on splicing process. In particular, the splice site variant c.2052+3G>T, detected in four unrelated families, resulted in skipping of exon 14 in DNAH5 transcripts and, according to haplotype analysis of affected probands, was proposed as an ancestral founder mutation in Udmurt population.
Conclusions: The reported data provide a vital insight into genetic background of primary ciliary dyskinesia in the Russian population. The findings clearly illustrate the utility of gene panel sequencing coupled with transcriptional analysis in identification and clinical interpretation of novel genetic variants.
Keywords: DNAH5; HYDIN; ZMYND10; Founder mutation; Gene-panel sequencing; Primary ciliary dyskinesia; Russian population; Splice site variants; Transcript analysis.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
