

Structural basis for allosteric regulation of human phosphofructokinase-1
- PMID: 39183237
- PMCID: PMC11345425
- DOI: 10.1038/s41467-024-51808-6
Structural basis for allosteric regulation of human phosphofructokinase-1
Abstract
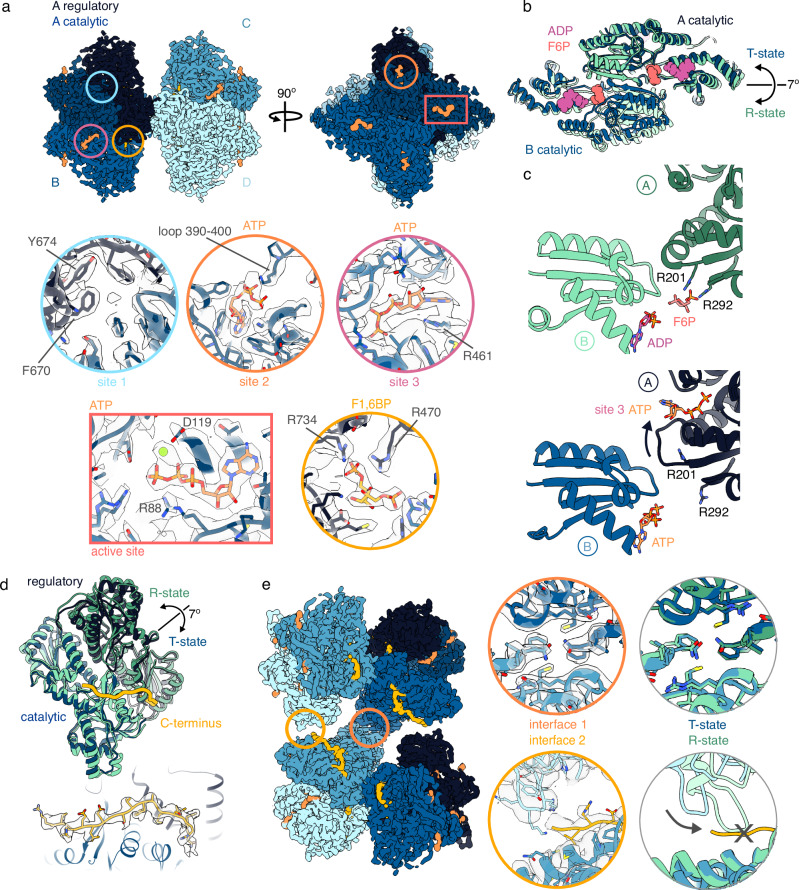
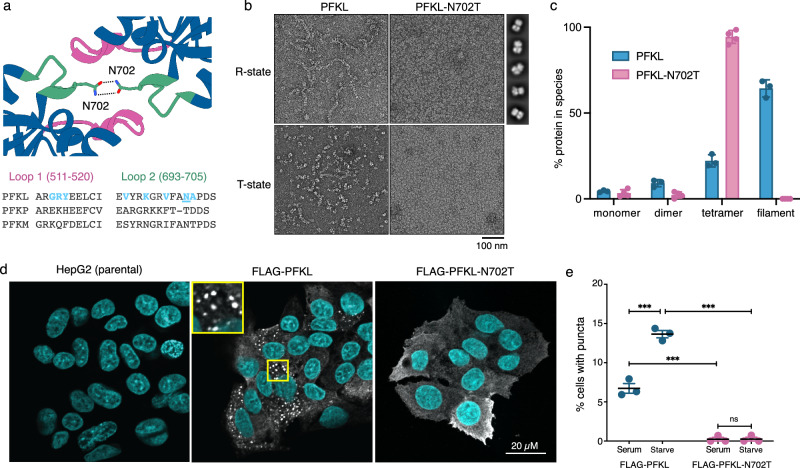
Phosphofructokinase-1 (PFK1) catalyzes the rate-limiting step of glycolysis, committing glucose to conversion into cellular energy. PFK1 is highly regulated to respond to the changing energy needs of the cell. In bacteria, the structural basis of PFK1 regulation is a textbook example of allostery; molecular signals of low and high cellular energy promote transition between an active R-state and inactive T-state conformation, respectively. Little is known, however, about the structural basis for regulation of eukaryotic PFK1. Here, we determine structures of the human liver isoform of PFK1 (PFKL) in the R- and T-state by cryoEM, providing insight into eukaryotic PFK1 allosteric regulatory mechanisms. The T-state structure reveals conformational differences between the bacterial and eukaryotic enzyme, the mechanisms of allosteric inhibition by ATP binding at multiple sites, and an autoinhibitory role of the C-terminus in stabilizing the T-state. We also determine structures of PFKL filaments that define the mechanism of higher-order assembly and demonstrate that these structures are necessary for higher-order assembly of PFKL in cells.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


Update of
-
Structural basis for allosteric regulation of human phosphofructokinase-1.bioRxiv [Preprint]. 2024 Mar 16:2024.03.15.585110. doi: 10.1101/2024.03.15.585110. bioRxiv. 2024. Update in: Nat Commun. 2024 Aug 25;15(1):7323. doi: 10.1038/s41467-024-51808-6. PMID: 38559074 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
- P20GM144230/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- F32 GM154453/GM/NIGMS NIH HHS/United States
- S10OD023476/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- S10 OD023476/OD/NIH HHS/United States
- P20 GM144230/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
