

A physics-aware neural network for protein-ligand interactions with quantum chemical accuracy
- PMID: 39183910
- PMCID: PMC11339967
- DOI: 10.1039/d4sc01029a
A physics-aware neural network for protein-ligand interactions with quantum chemical accuracy
Abstract
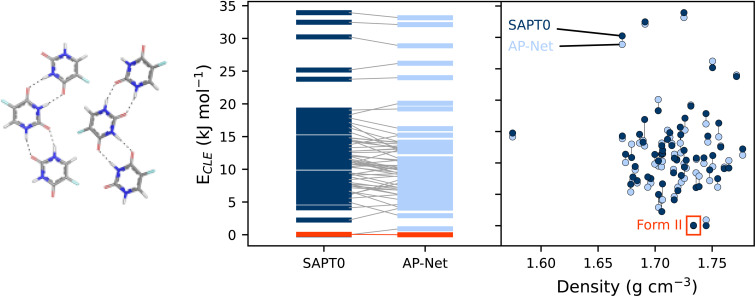
Quantifying intermolecular interactions with quantum chemistry (QC) is useful for many chemical problems, including understanding the nature of protein-ligand interactions. Unfortunately, QC computations on protein-ligand systems are too computationally expensive for most use cases. The flourishing field of machine-learned (ML) potentials is a promising solution, but it is limited by an inability to easily capture long range, non-local interactions. In this work we develop an atomic-pairwise neural network (AP-Net) specialized for modeling intermolecular interactions. This model benefits from a number of physical constraints, including a two-component equivariant message passing neural network architecture that predicts interaction energies via an intermediate prediction of monomer electron densities. The AP-Net model is trained on a comprehensive dataset composed of paired ligand and protein fragments. This model accurately predicts QC-quality interaction energies of protein-ligand systems at a computational cost reduced by orders of magnitude. Applications of the AP-Net model to molecular crystal structure prediction are explored, as well as limitations in modeling highly polarizable systems.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures





References
-
- Bodley A. Liu L. F. Israel M. Seshadri R. Koseki Y. Giuliani F. C. Kirschenbaum S. Silber R. Potmesil M. Cancer Res. 1989;49:5969–5978. - PubMed
LinkOut - more resources
Full Text Sources