

This is a preprint.
The Aryl Hydrocarbon Receptor Controls IFNγ-Induced Immune Checkpoints PD-L1 and IDO via the JAK/STAT Pathway in Lung Adenocarcinoma
- PMID: 39185148
- PMCID: PMC11343147
- DOI: 10.1101/2024.08.12.607602
The Aryl Hydrocarbon Receptor Controls IFNγ-Induced Immune Checkpoints PD-L1 and IDO via the JAK/STAT Pathway in Lung Adenocarcinoma
Update in
-
The aryl hydrocarbon receptor controls IFN-γ-induced immune checkpoints PD-L1 and IDO via the JAK/STAT pathway in lung adenocarcinoma.J Immunol. 2025 Mar 1;214(3):413-432. doi: 10.1093/jimmun/vkae023. J Immunol. 2025. PMID: 40073102
Abstract
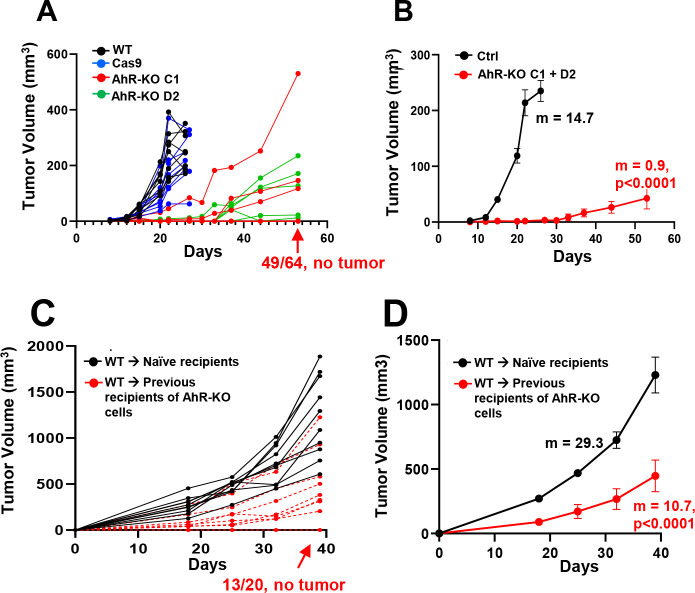
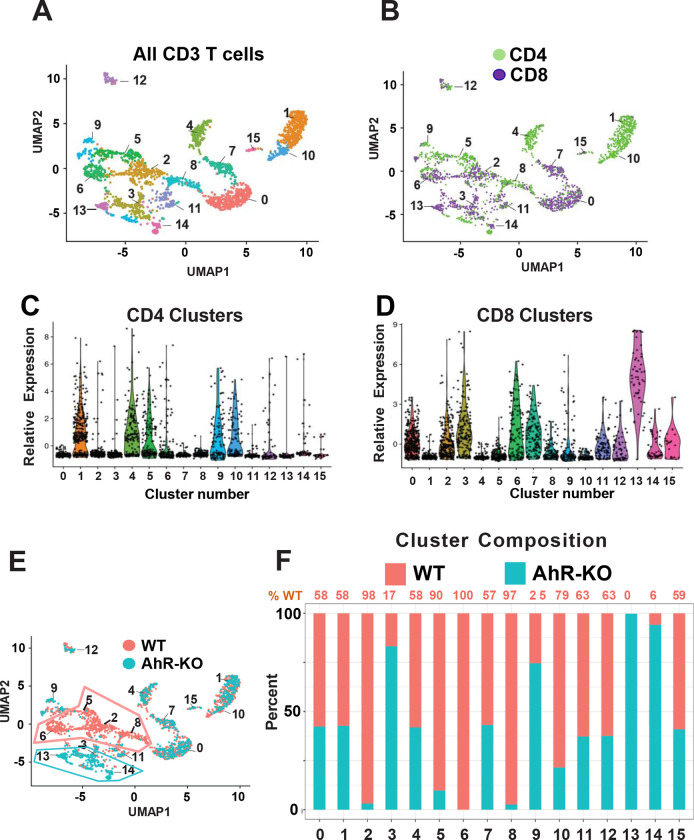
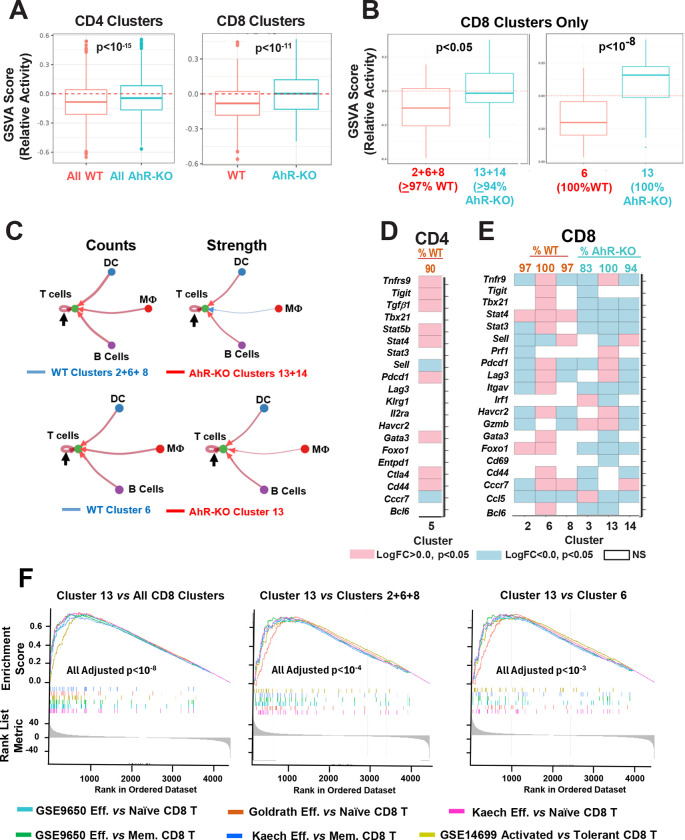
While immunotherapy has shown efficacy in lung adenocarcinoma (LUAD) patients, many respond only partially or not at all. One limitation in improving outcomes is the lack of a complete understanding of immune checkpoint regulation. Here, we investigated a possible link between an environmental chemical receptor implicated in lung cancer and immune regulation, (the aryl hydrocarbon receptor/AhR), a known but counterintuitive mediator of immunosuppression (IFNγ), and regulation of two immune checkpoints (PD-L1 and IDO). AhR gene-edited LUAD cell lines, a syngeneic LUAD mouse model, bulk- and scRNA sequencing of LUADs and tumor-infiltrating leukocytes were used to map out a signaling pathway leading from IFNγ through the AhR to JAK/STAT, PD-L1, IDO, and tumor-mediated immunosuppression. The data demonstrate that: 1) IFNγ activation of the JAK/STAT pathway leading to PD-L1 and IDO1 upregulation is mediated by the AhR in murine and human LUAD cells, 2) AhR-driven IDO1 induction results in the production of Kynurenine (Kyn), an AhR ligand, which likely mediates an AhR→IDO1→Kyn→AhR amplification loop, 3) transplantation of AhR-knockout LUAD cells results in long-term tumor immunity in most recipients. 4) The 23% of AhR-knockout tumors that do grow do so at a much slower pace than controls and exhibit higher densities of CD8+ T cells expressing markers of immunocompetence, increased activity, and increased cell-cell communication. The data definitively link the AhR to IFNγ-induced JAK/STAT pathway and immune checkpoint-mediated immunosuppression and support the targeting of the AhR in the context of LUAD.
Keywords: Aryl Hydrocarbon Receptor; Cancer; Interferon Gamma; Tumor Immunity.
Figures







References
-
- Ferlay J., Shin H. R., Bray F., Forman D., Mathers C., and Parkin D. M.. 2010. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127: 2893–2917. - PubMed
-
- Rivera G. A., and Wakelee H.. 2016. Lung Cancer in Never Smokers. Adv Exp Med Biol 893: 43–57. - PubMed
-
- Sezer A., Kilickap S., Gumus M., Bondarenko I., Ozguroglu M., Gogishvili M., Turk H. M., Cicin I., Bentsion D., Gladkov O., Clingan P., Sriuranpong V., Rizvi N., Gao B., Li S., Lee S., McGuire K., Chen C. I., Makharadze T., Paydas S., Nechaeva M., Seebach F., Weinreich D. M., Yancopoulos G. D., Gullo G., Lowy I., and Rietschel P.. 2021. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet 397: 592–604. - PubMed
-
- Paz-Ares L. G., Ramalingam S. S., Ciuleanu T. E., Lee J. S., Urban L., Caro R. B., Park K., Sakai H., Ohe Y., Nishio M., Audigier-Valette C., Burgers J. A., Pluzanski A., Sangha R., Gallardo C., Takeda M., Linardou H., Lupinacci L., Lee K. H., Caserta C., Provencio M., Carcereny E., Otterson G. A., Schenker M., Zurawski B., Alexandru A., Vergnenegre A., Raimbourg J., Feeney K., Kim S. W., Borghaei H., O'Byrne K. J., Hellmann M. D., Memaj A., Nathan F. E., Bushong J., Tran P., Brahmer J. R., and Reck M.. 2022. First-Line Nivolumab Plus Ipilimumab in Advanced NSCLC: 4-Year Outcomes From the Randomized, Open-Label, Phase 3 CheckMate 227 Part 1 Trial. J Thorac Oncol 17: 289–308. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials