

This is a preprint.
Identifying a gene signature of metastatic potential by linking pre-metastatic state to ultimate metastatic fate
- PMID: 39185156
- PMCID: PMC11343111
- DOI: 10.1101/2024.08.14.607813
Identifying a gene signature of metastatic potential by linking pre-metastatic state to ultimate metastatic fate
Update in
-
A cell-state axis underlying colonization in carcinomas with implications for metastasis risk prediction and interception.Cell Rep. 2025 Dec 23;44(12):116701. doi: 10.1016/j.celrep.2025.116701. Epub 2025 Dec 15. Cell Rep. 2025. PMID: 41401069 Free PMC article.
Abstract
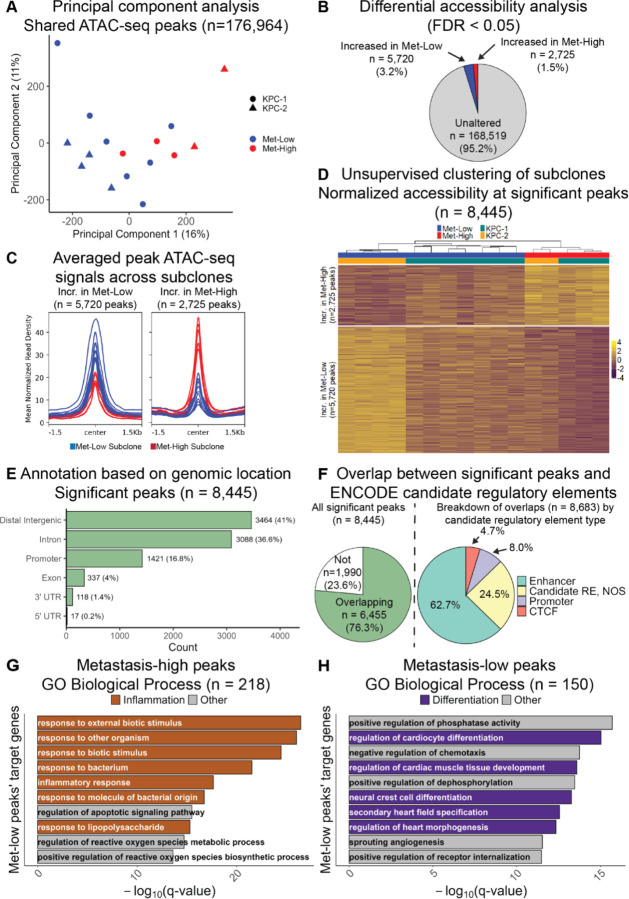
Identifying the key molecular pathways that enable metastasis by analyzing the eventual metastatic tumor is challenging because the state of the founder subclone likely changes following metastatic colonization. To address this challenge, we labeled primary mouse pancreatic ductal adenocarcinoma (PDAC) subclones with DNA barcodes to characterize their pre-metastatic state using ATAC-seq and RNA-seq and determine their relative in vivo metastatic potential prospectively. We identified a gene signature separating metastasis-high and metastasis-low subclones orthogonal to the normal-to-PDAC and classical-to-basal axes. The metastasis-high subclones feature activation of IL-1 pathway genes and high NF-κB and Zeb/Snail family activity and the metastasis-low subclones feature activation of neuroendocrine, motility, and Wnt pathway genes and high CDX2 and HOXA13 activity. In a functional screen, we validated novel mediators of PDAC metastasis in the IL-1 pathway, including the NF-κB targets Fos and Il23a, and beyond the IL-1 pathway including Myo1b and Tmem40. We scored human PDAC tumors for our signature of metastatic potential from mouse and found that metastases have higher scores than primary tumors. Moreover, primary tumors with higher scores are associated with worse prognosis. We also found that our metastatic potential signature is enriched in other human carcinomas, suggesting that it is conserved across epithelial malignancies. This work establishes a strategy for linking cancer cell state to future behavior, reveals novel functional regulators of PDAC metastasis, and establishes a method for scoring human carcinomas based on metastatic potential.
Conflict of interest statement
Competing Financial Interests J.S.H. and R.K. are listed as co-inventors on a provisional patent on the use of this study’s findings for clinical metastasis risk prediction. E.J.F serves on the Scientific Advisory Board of Resistance Bio, as a consultant for Mestag Therapeutics and Merck, and receives research funding from Abbvie Inc and Roche/Genetech outside the scope of this work.
Figures






References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials