Birth of protein folds and functions in the virome
- PMID: 39187718
- PMCID: PMC11410667
- DOI: 10.1038/s41586-024-07809-y
Birth of protein folds and functions in the virome
Erratum in
-
Author Correction: Birth of protein folds and functions in the virome.Nature. 2024 Oct;634(8033):E4. doi: 10.1038/s41586-024-08068-7. Nature. 2024. PMID: 39313532 Free PMC article. No abstract available.
Abstract
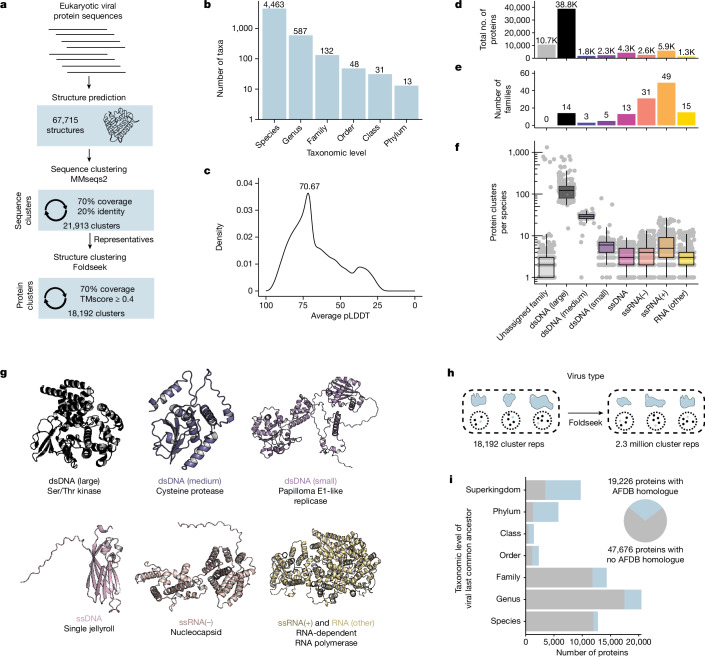
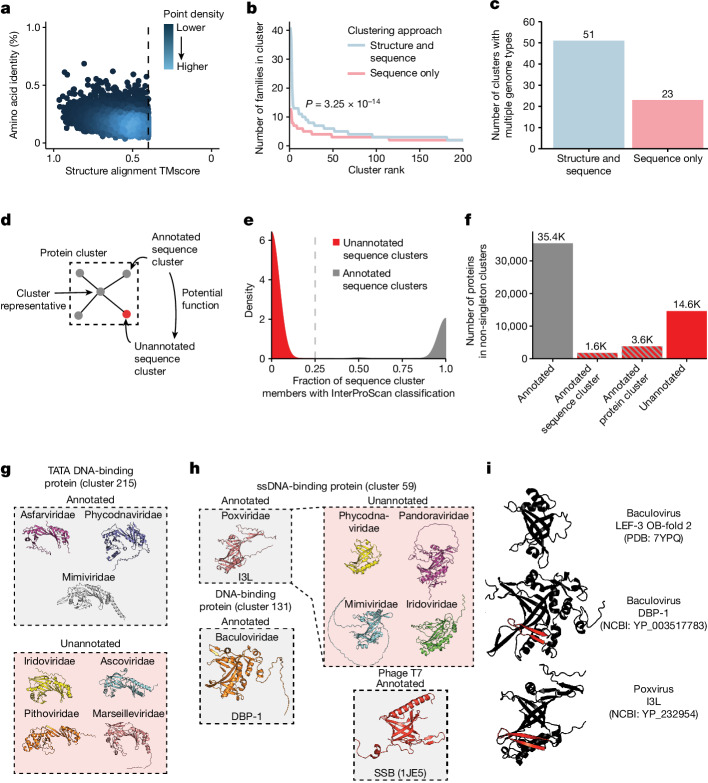
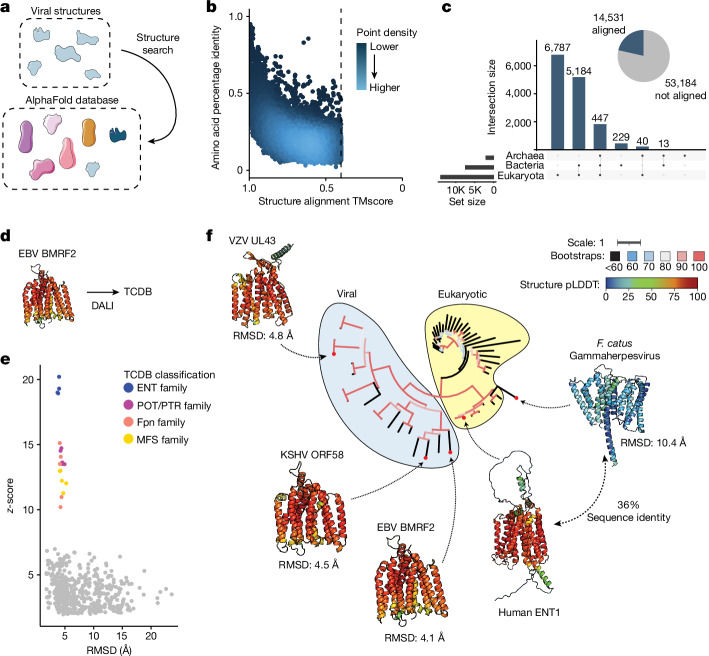
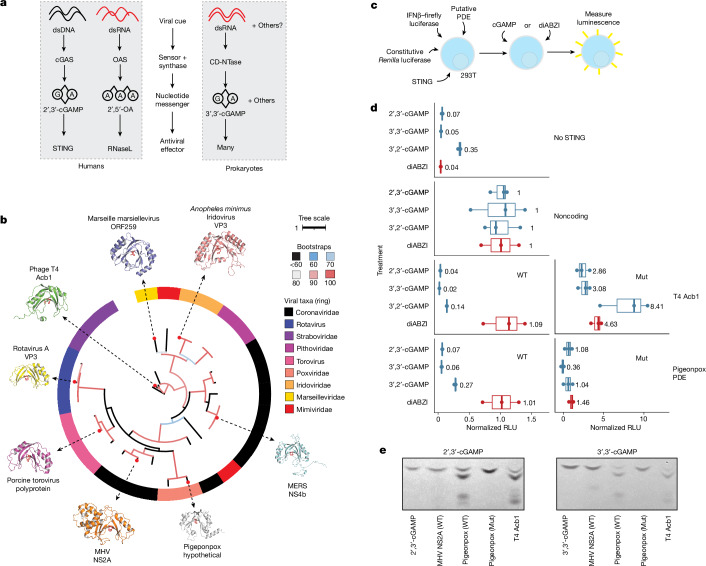
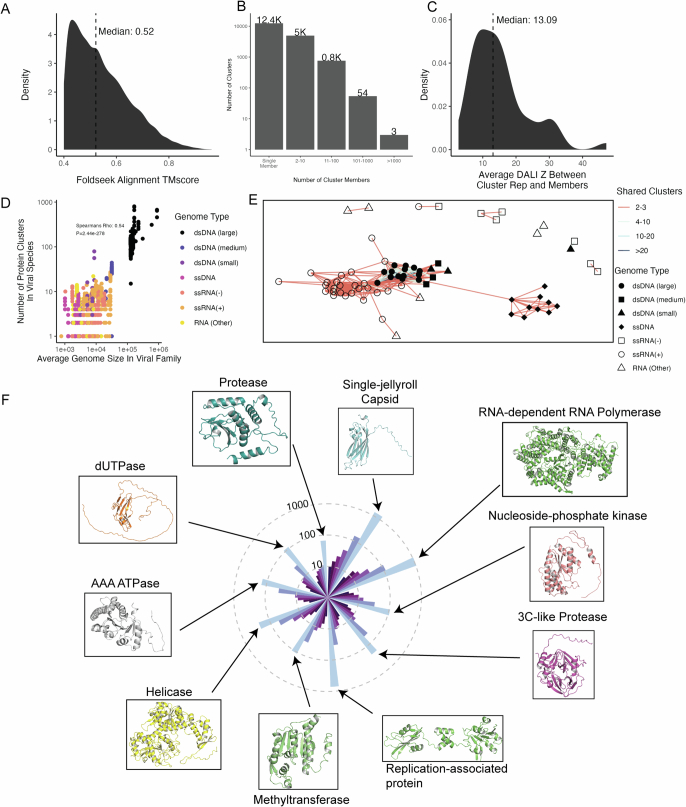
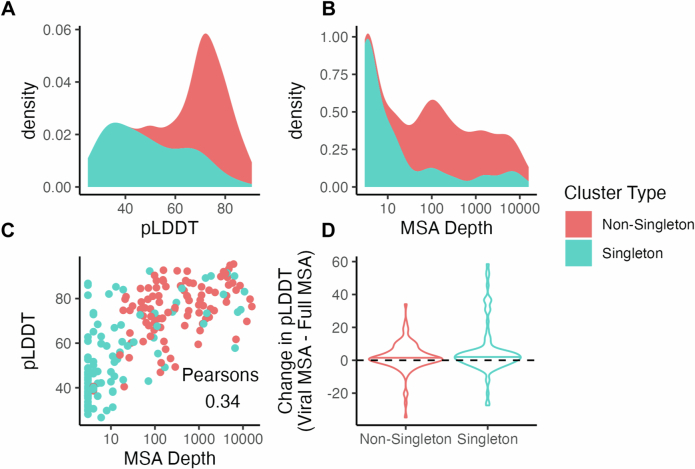
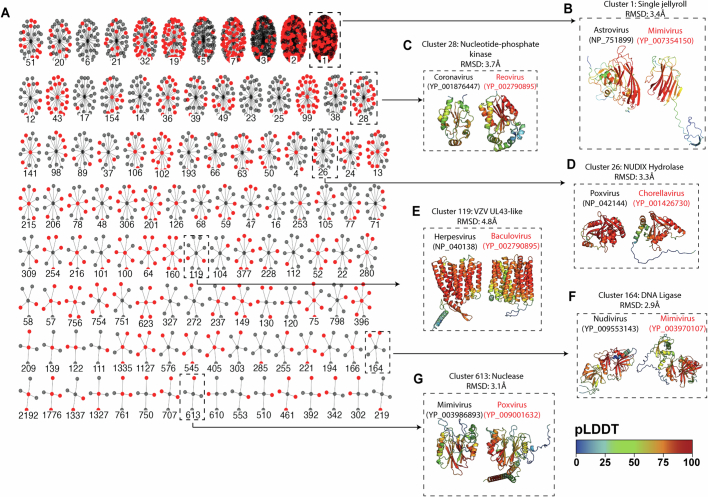
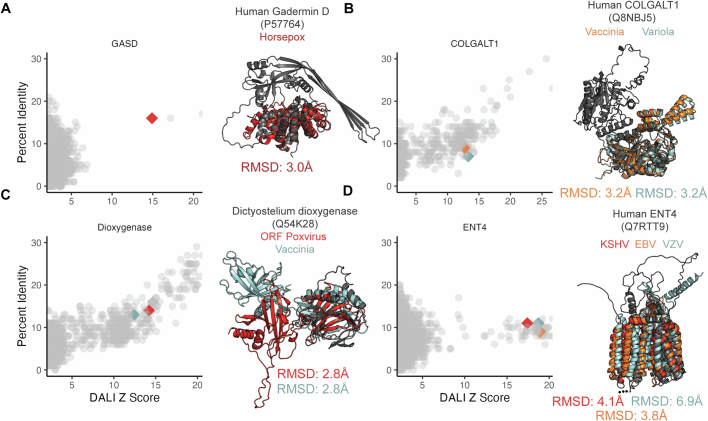
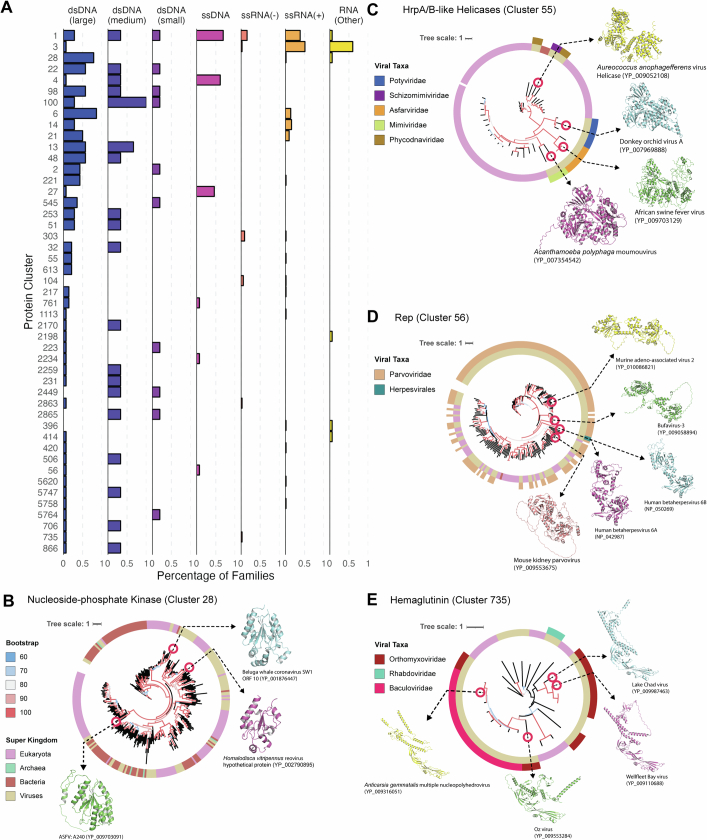
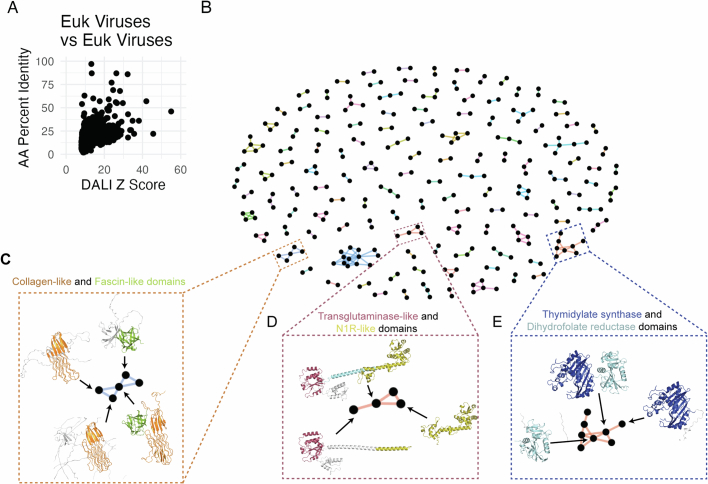
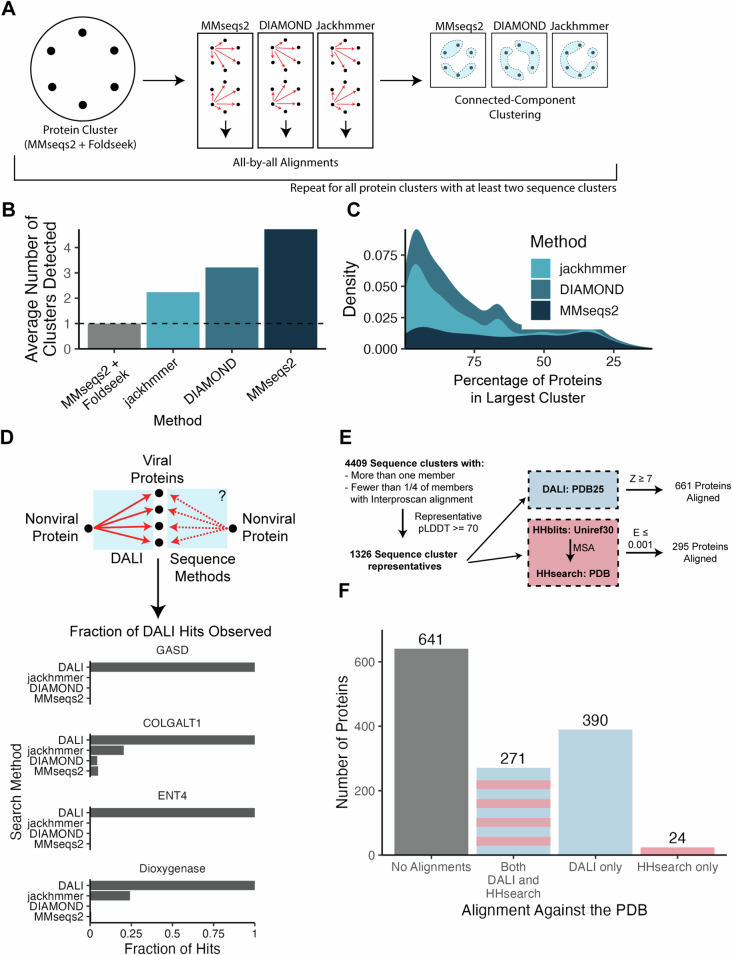
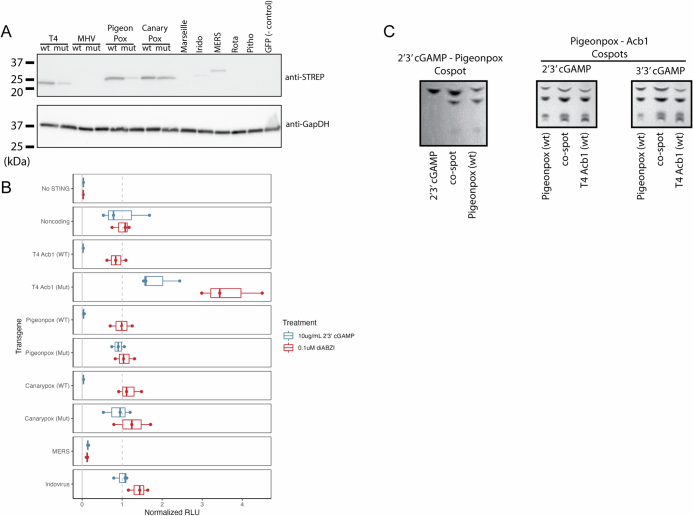
The rapid evolution of viruses generates proteins that are essential for infectivity and replication but with unknown functions, due to extreme sequence divergence1. Here, using a database of 67,715 newly predicted protein structures from 4,463 eukaryotic viral species, we found that 62% of viral proteins are structurally distinct and lack homologues in the AlphaFold database2,3. Among the remaining 38% of viral proteins, many have non-viral structural analogues that revealed surprising similarities between human pathogens and their eukaryotic hosts. Structural comparisons suggested putative functions for up to 25% of unannotated viral proteins, including those with roles in the evasion of innate immunity. In particular, RNA ligase T-like phosphodiesterases were found to resemble phage-encoded proteins that hydrolyse the host immune-activating cyclic dinucleotides 3',3'- and 2',3'-cyclic GMP-AMP (cGAMP). Experimental analysis showed that RNA ligase T homologues encoded by avian poxviruses similarly hydrolyse cGAMP, showing that RNA ligase T-mediated targeting of cGAMP is an evolutionarily conserved mechanism of immune evasion that is present in both bacteriophage and eukaryotic viruses. Together, the viral protein structural database and analyses presented here afford new opportunities to identify mechanisms of virus-host interactions that are common across the virome.
© 2024. The Author(s).
Conflict of interest statement
The Regents of the University of California have patents issued and pending for CRISPR technologies on which J.A.D. is an inventor. J.A.D. and J.N. are listed as inventors on a patent filing related to DNA-binding proteins characterized in this work. J.A.D. is a cofounder of Azalea Therapeutics, Caribou Biosciences, Editas Medicine, Evercrisp, Scribe Therapeutics, Intellia Therapeutics and Mammoth Biosciences. J.A.D. is a scientific advisory board member at Evercrisp, Caribou Biosciences, Intellia Therapeutics, Scribe Therapeutics, Mammoth Biosciences, The Column Group and Inari. She also is an advisor for Aditum Bio. J.A.D. is Chief Science Advisor to Sixth Street, a Director at Johnson & Johnson, Altos and Tempus, and has research projects sponsored by Apple Tree Partners. The other authors declare no competing interests.
Figures