

A novel deletion in the BLOC1S6 Gene Associated with Hermansky-Pudlak syndrome type 9 (HPS-9)
- PMID: 39187771
- PMCID: PMC11348666
- DOI: 10.1186/s12864-024-10478-w
A novel deletion in the BLOC1S6 Gene Associated with Hermansky-Pudlak syndrome type 9 (HPS-9)
Abstract
Background: Hermansky-Pudlak Syndrome (HPS), a rare autosomal recessive disorder, is characterized by oculocutaneous albinism, bleeding diathesis, and sometimes severe lung problems and inflammatory bowel disease. Symptoms include skin and hair pigmentation variations, along with visual impairments. Variants in eleven genes encoding protein complexes essential for membrane trafficking and intracellular endosomal transport pathways underlie various recognized HPS subtypes. This study focuses on HPS-9, a subtype of Hermansky-Pudlak Syndrome caused by a variant in the BLOC1S6 gene, which is a subunit of the BLOC1 complex. In this study, a novel Copy Number Variation (CNV) in the aforementioned gene in an Iranian family is reported. The study aims to better understand the etiology of HPS-9 symptoms by identifying and confirming the variant and determining whether the gene is expressed despite the deletion. There have only been five reports of this syndrome in the literature thus far. Our novel CNV represents a significant contribution to understanding the genetic basis of HPS-9.
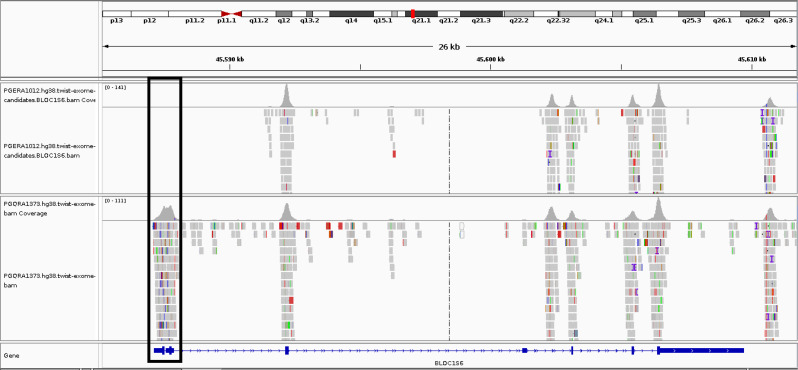
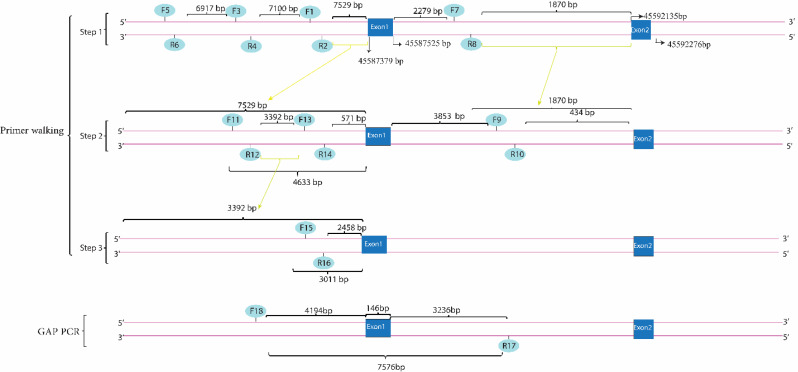
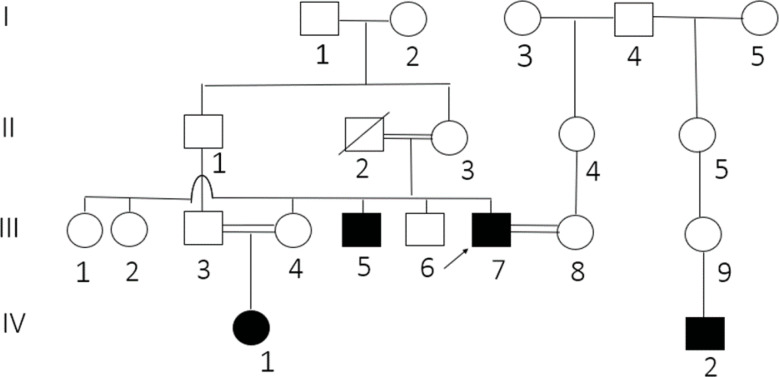
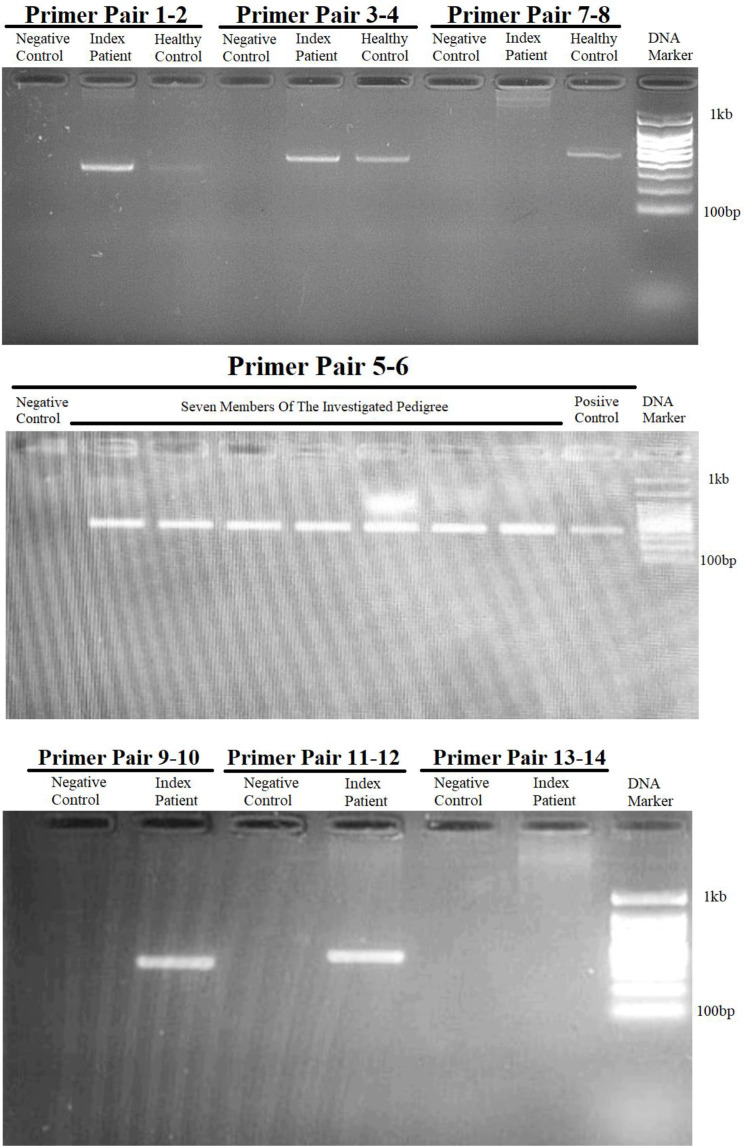
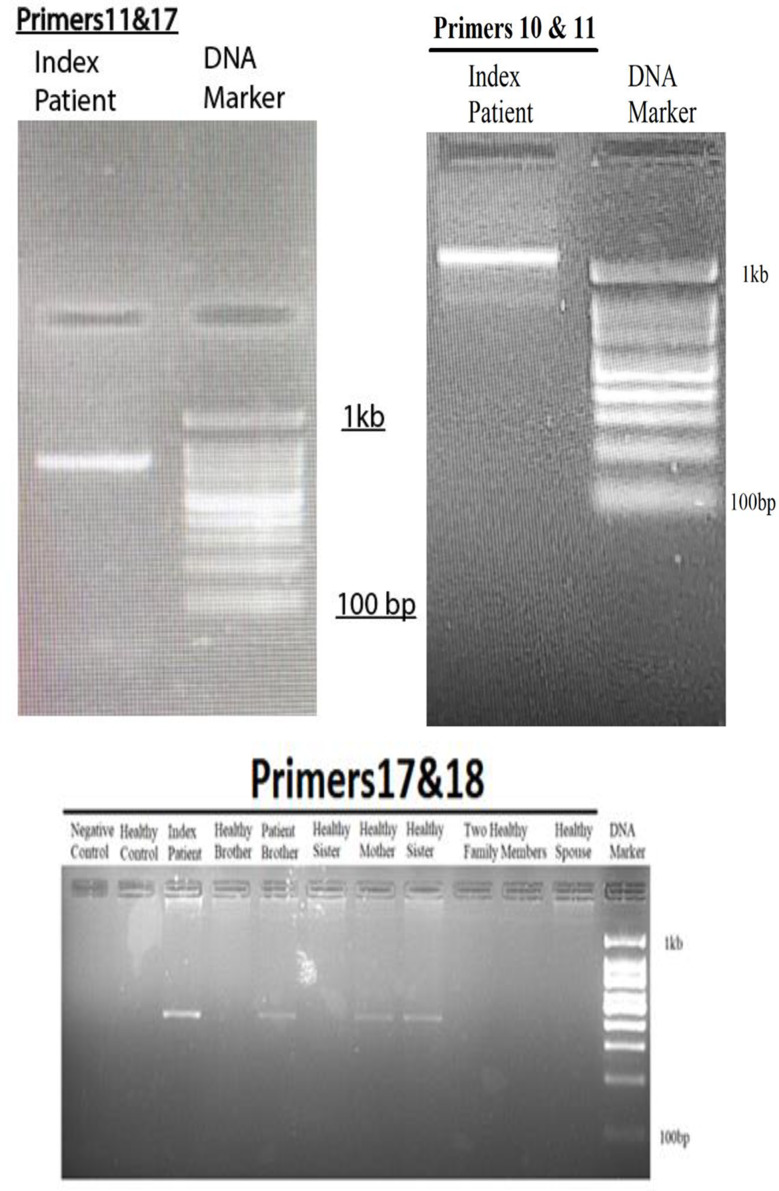
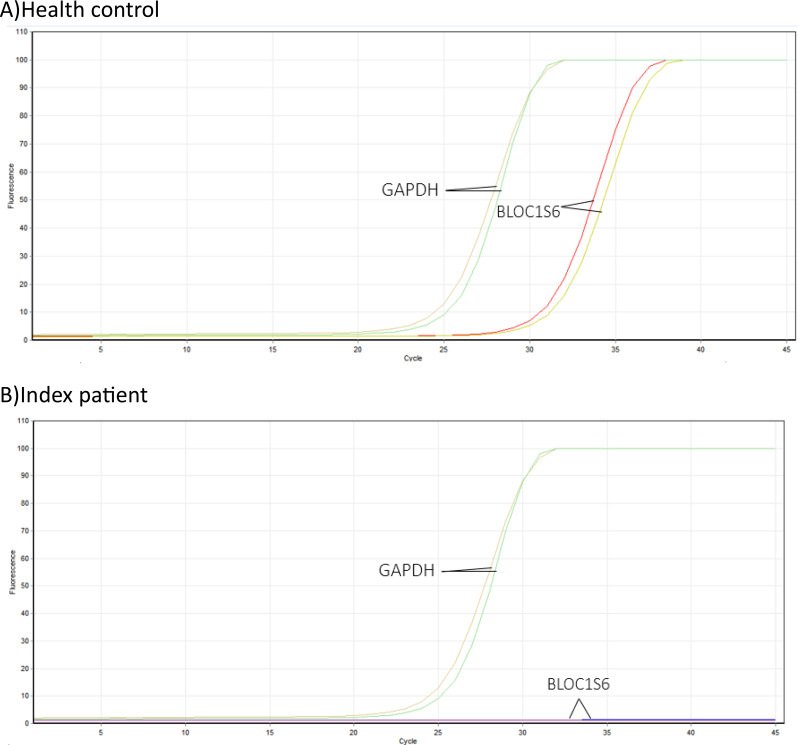
Results: This study investigates a male patient presenting with albinism. Whole Exome Sequencing (WES) identified a homozygous deletion of approximately 350 bp using CNV analysis. The deletion affects the intronic region of the BLOC1S6 gene, causing uncertainties in defining the exact boundaries due to WES limitations. Primer walking and GAP-PCR techniques were used to define the deletion boundaries. Subsequent assessments of this variant across other family members helped identify homozygous affected members and heterozygous carriers. The absence of BLOC1S6 expression in the affected individual was confirmed through Real-time PCR experiments. These findings underscore the importance of understanding the implications for the patient's healthcare and potential therapeutic approaches.
Conclusion: This study introduces a case of Hermansky-Pudlak Syndrome Type 9 (HPS-9) caused by a homozygous deletion in the BLOC1S6 gene. We identified an approximately 7-kb deletion encompassing exon 1 and the intronic region of the gene. The absence of BLOC1S6 expression, confirmed via Real-time PCR, highlights the importance of studying the pathogenicity of the deletion and its impact on the patient's health. Our findings contribute to the sparse knowledge on HPS-9 and underscore the need for further exploration into the genetic causes of this rare disorder.
Keywords: Albinism; GAP-PCR; Hermansky-pudlock syndrome type 9; Primer-walking; Whole exome sequencing.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
