

A single-cell atlas of the miracidium larva of Schistosoma mansoni reveals cell types, developmental pathways, and tissue architecture
- PMID: 39190022
- PMCID: PMC11349301
- DOI: 10.7554/eLife.95628
A single-cell atlas of the miracidium larva of Schistosoma mansoni reveals cell types, developmental pathways, and tissue architecture
Abstract
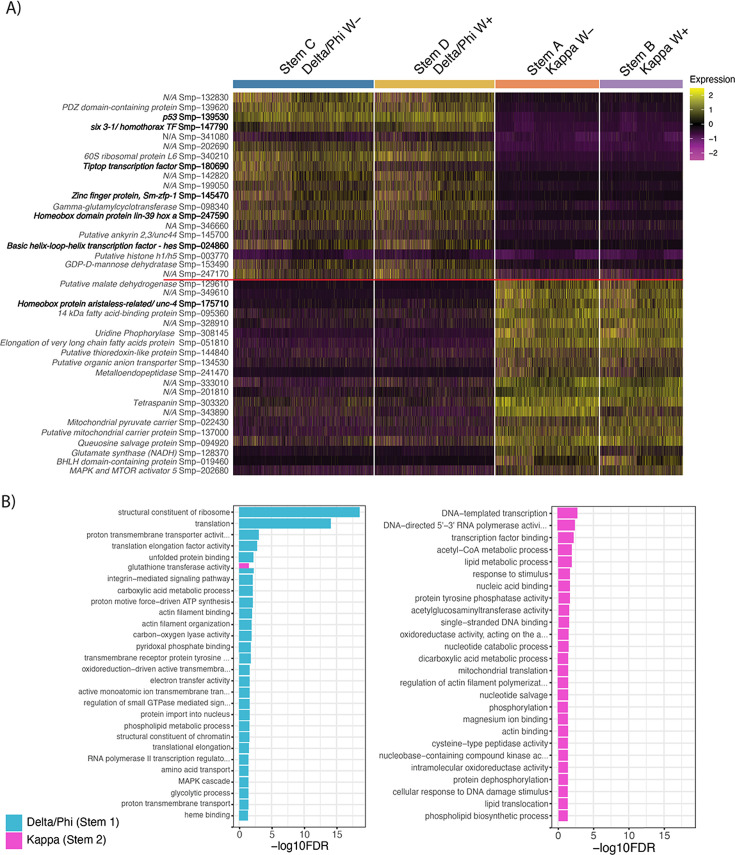
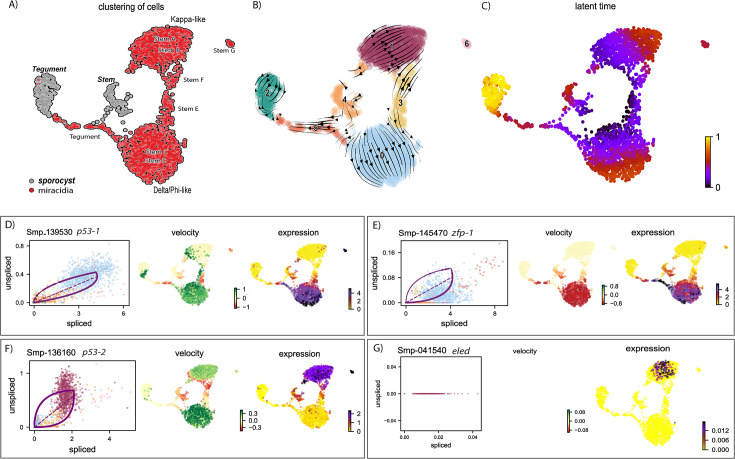
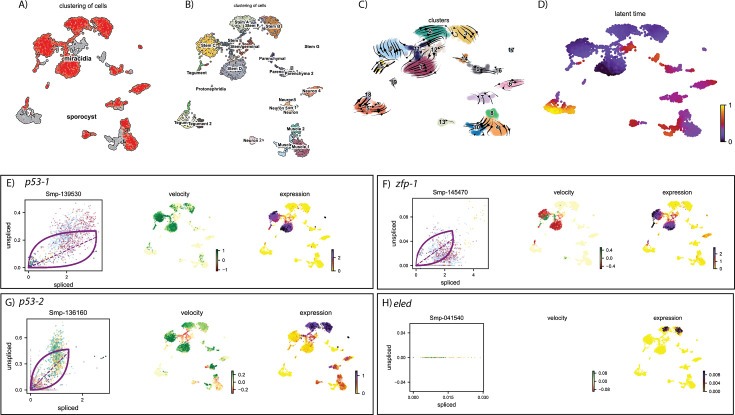
Schistosoma mansoni is a parasitic flatworm that causes the major neglected tropical disease schistosomiasis. The miracidium is the first larval stage of the life cycle. It swims and infects a freshwater snail, transforms into a mother sporocyst, where its stem cells generate daughter sporocysts that give rise to human-infective cercariae larvae. To understand the miracidium at cellular and molecular levels, we created a whole-body atlas of its ~365 cells. Single-cell RNA sequencing identified 19 transcriptionally distinct cell clusters. In situ hybridisation of tissue-specific genes revealed that 93% of the cells in the larva are somatic (57% neural, 19% muscle, 13% epidermal or tegument, 2% parenchyma, and 2% protonephridia) and 7% are stem. Whereas neurons represent the most diverse somatic cell types, trajectory analysis of the two main stem cell populations indicates that one of them is the origin of the tegument lineage and the other likely contains pluripotent cells. Furthermore, unlike the somatic cells, each of these stem populations shows sex-biased transcriptional signatures suggesting a cell-type-specific gene dosage compensation for sex chromosome-linked loci. The miracidium represents a simple developmental stage with which to gain a fundamental understanding of the molecular biology and spatial architecture of schistosome cells.
Keywords: Schistosoma mansoni; genetics; genomics; infectious disease; microbiology; miracidium; single-cell atlas; single-cell sequencing.
© 2024, Attenborough, Rawlinson et al.
Conflict of interest statement
TA, KR, CD, KA, GS, JG, JC, SD, GR, MB No competing interests declared
Figures






























Update of
- doi: 10.1101/2023.03.27.533868
- doi: 10.7554/eLife.95628.1
- doi: 10.7554/eLife.95628.2
References
-
- Alexa A, Rahnenfuhrer J. topGO: Enrichment Analysis for Gene Ontology. version 2.54.0R Package. 2023 https://bioconductor.org/packages/release/bioc/html/topGO.html
-
- Attenborough T. Singlecell-miracidia. swh:1:rev:77941278d9ddd01cf41dcf556a7cf8dd00c97bbdSoftware Heritage. 2024 https://archive.softwareheritage.org/swh:1:dir:c80776b59598432be422eb243...
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
