

Ecological Changes Exacerbating the Spread of Invasive Ticks has Driven the Dispersal of Severe Fever with Thrombocytopenia Syndrome Virus Throughout Southeast Asia
- PMID: 39191515
- PMCID: PMC11349436
- DOI: 10.1093/molbev/msae173
Ecological Changes Exacerbating the Spread of Invasive Ticks has Driven the Dispersal of Severe Fever with Thrombocytopenia Syndrome Virus Throughout Southeast Asia
Abstract
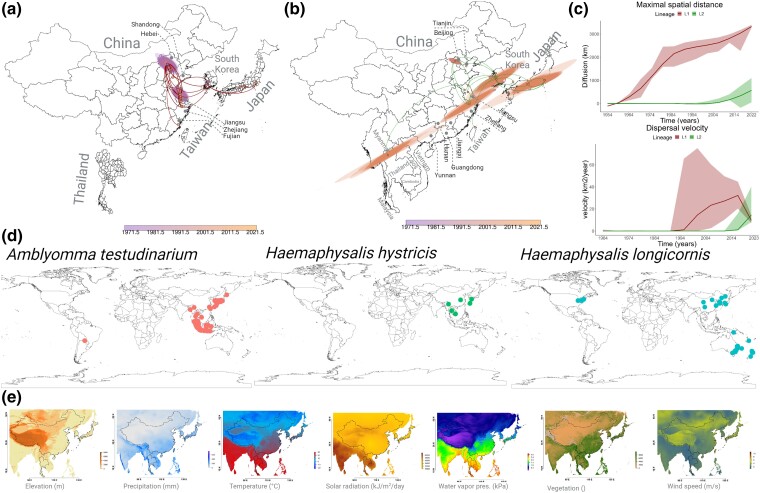
Severe fever with thrombocytopenia syndrome virus (SFTSV) is a tick-borne virus recognized by the World Health Organization as an emerging infectious disease of growing concern. Utilizing phylodynamic and phylogeographic methods, we have reconstructed the origin and transmission patterns of SFTSV lineages and the roles demographic, ecological, and climatic factors have played in shaping its emergence and spread throughout Asia. Environmental changes and fluctuations in tick populations, exacerbated by the widespread use of pesticides, have contributed significantly to its geographic expansion. The increased adaptability of Lineage L2 strains to the Haemaphysalis longicornis vector has facilitated the dispersal of SFTSV through Southeast Asia. Increased surveillance and proactive measures are needed to prevent further spread to Australia, Indonesia, and North America.
Keywords: climatic factors; ecological; evolution; phylodynamic; severe fever with thrombocytopenia syndrome virus.
© The Author(s) 2024. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
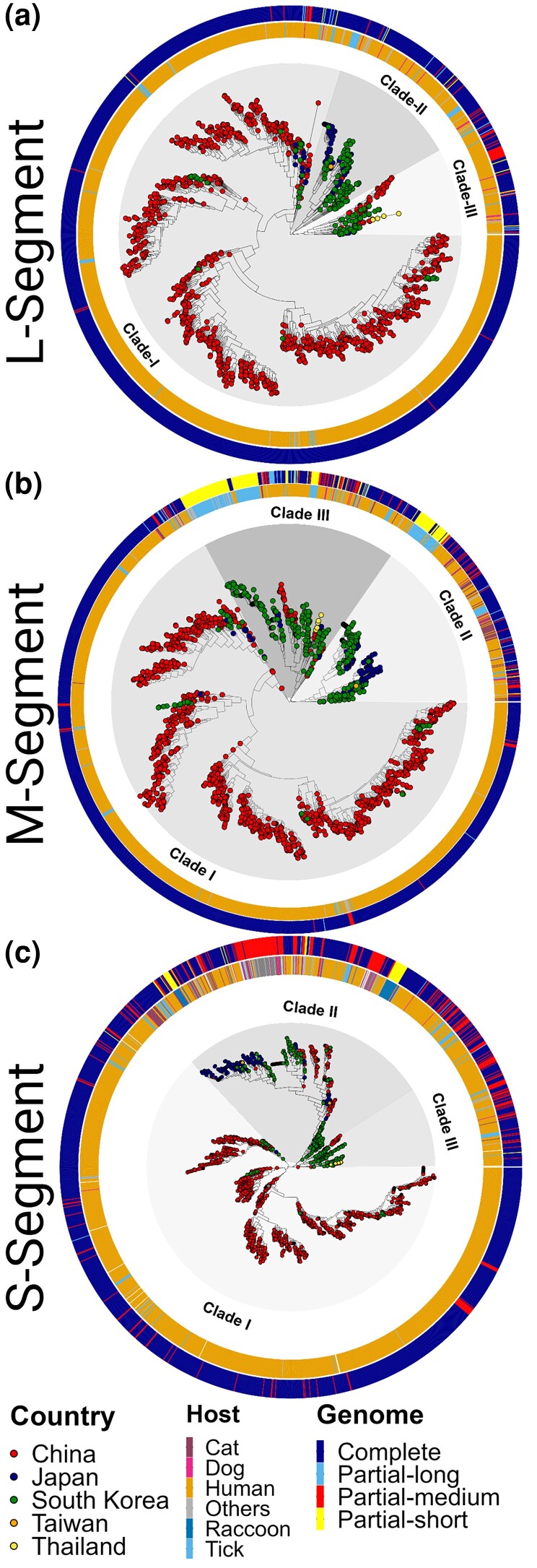
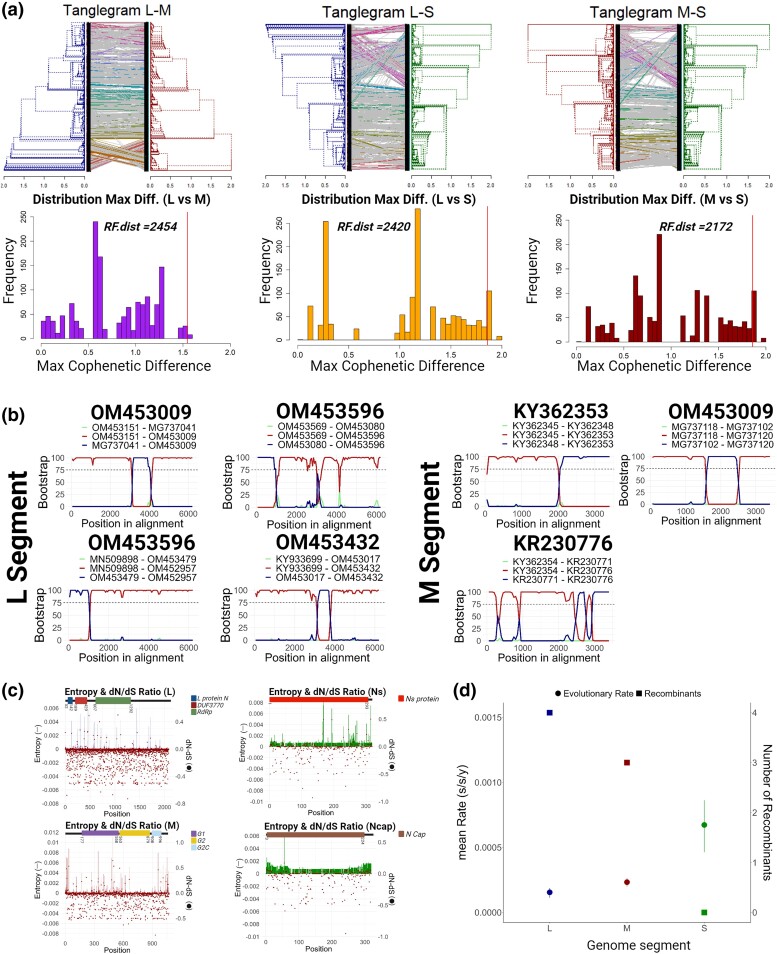
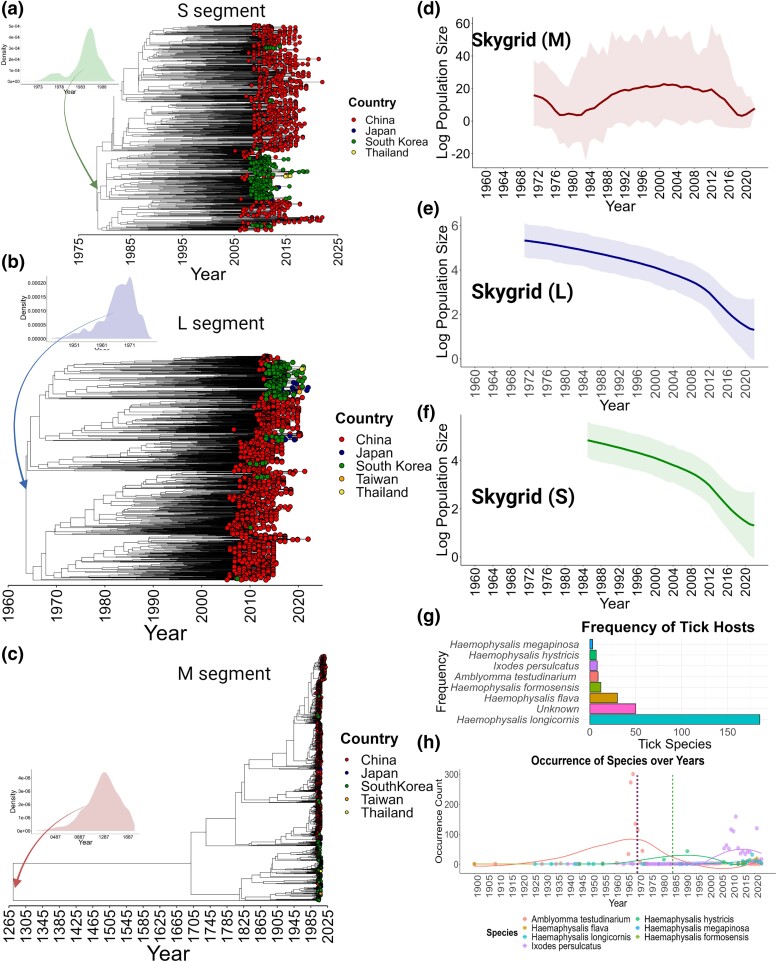
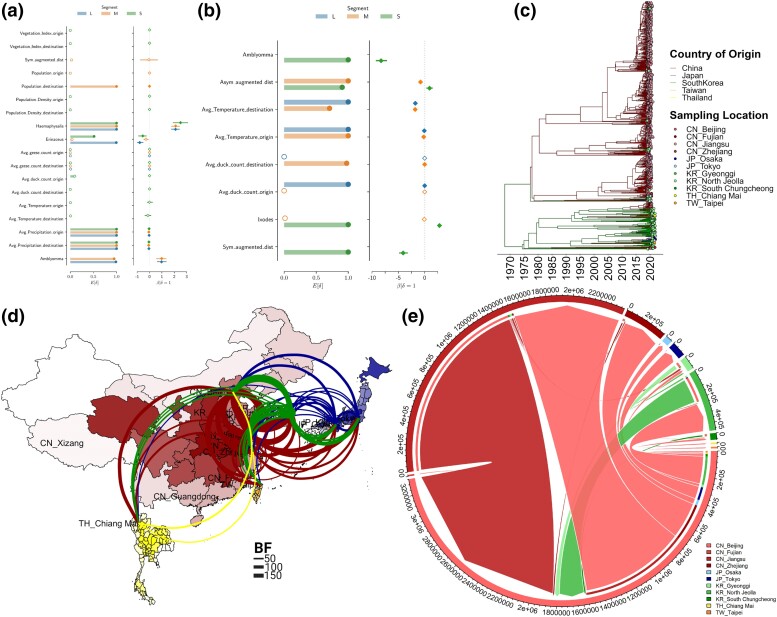
References
-
- Akagi K, Miyazaki T, Oshima K, Umemura A, Shimada S, Morita K, Senju H, Tashiro M, Takazono T, Saijo T, et al. . Detection of viral RNA in diverse body fluids in an SFTS patient with encephalopathy, gastrointestinal bleeding and pneumonia: a case report and literature review. BMC Infect Dis. 2020:20(1):281. 10.1186/s12879-020-05012-8. - DOI - PMC - PubMed
-
- Ayres DL, Cummings MP, Baele G, Darling AE, Lewis PO, Swofford DL, Huelsenbeck JP, Lemey P, Rambaut A, Suchard MA. BEAGLE 3: improved performance, scaling, and usability for a high-performance computing library for statistical phylogenetics. Syst Biol. 2019:68(6):1052–1061. 10.1093/sysbio/syz020. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
