

Genetic architecture of telomere length in 462,666 UK Biobank whole-genome sequences
- PMID: 39192095
- PMCID: PMC11387196
- DOI: 10.1038/s41588-024-01884-7
Genetic architecture of telomere length in 462,666 UK Biobank whole-genome sequences
Abstract
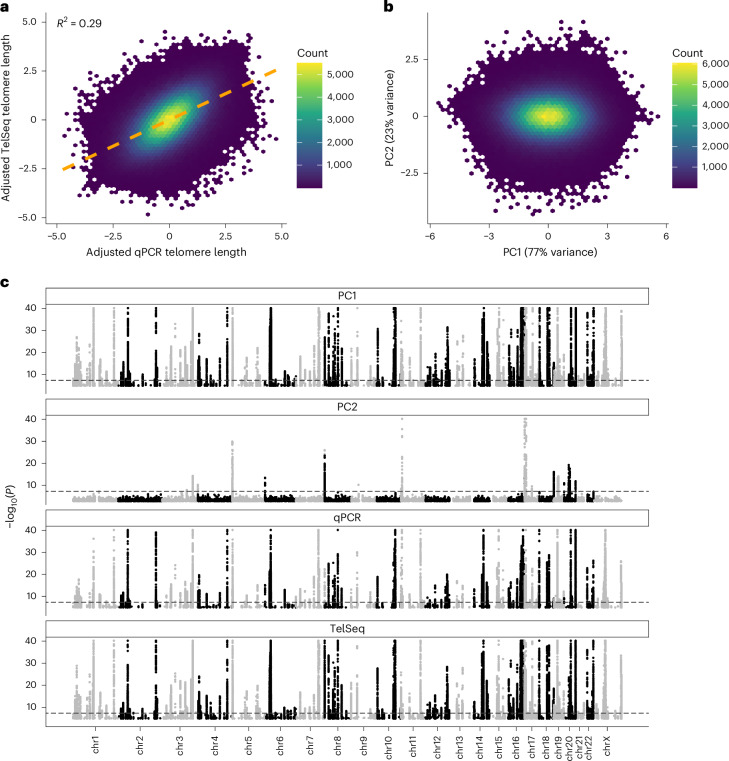
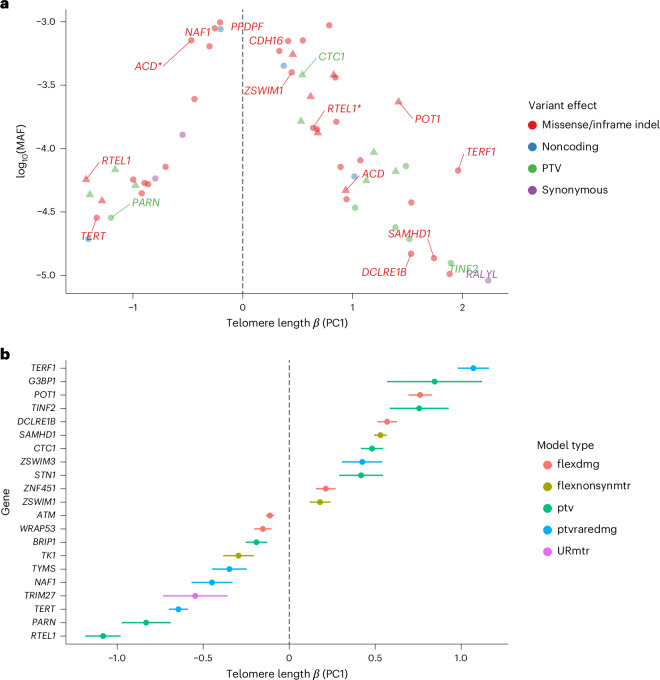
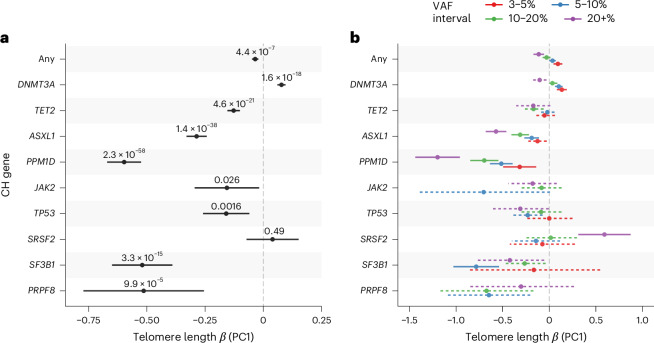
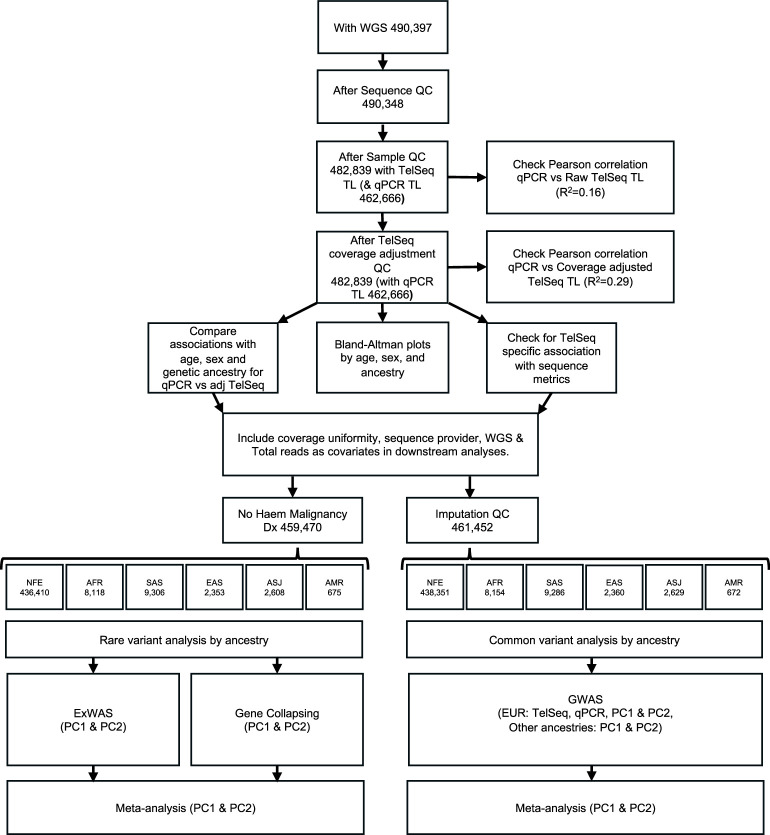
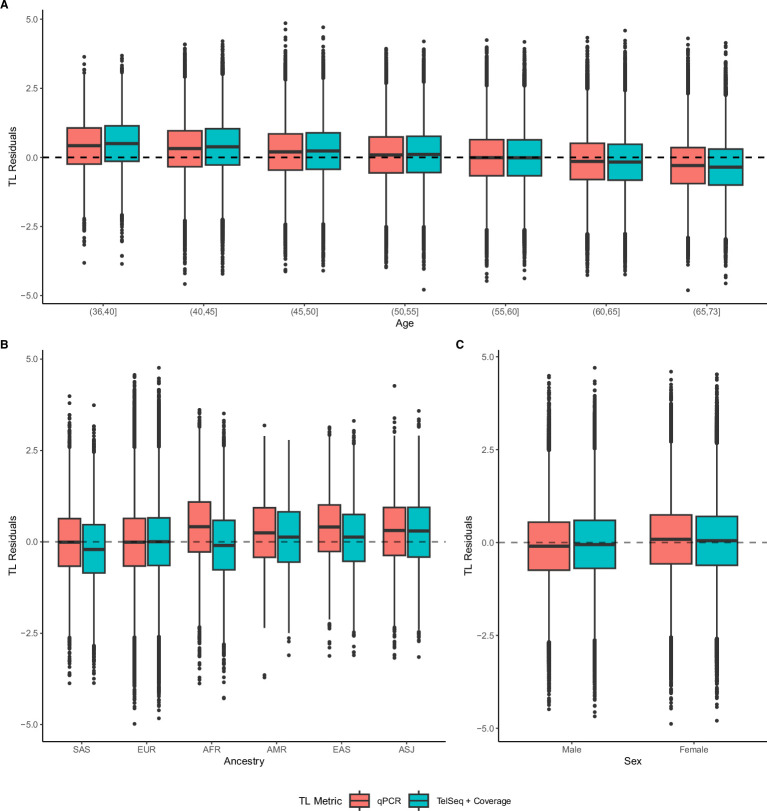
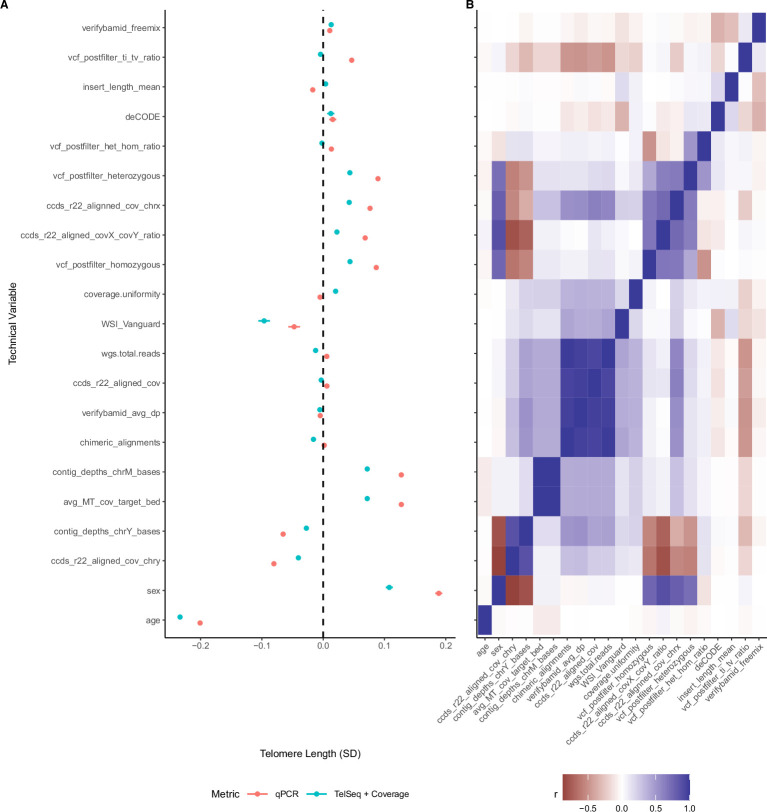
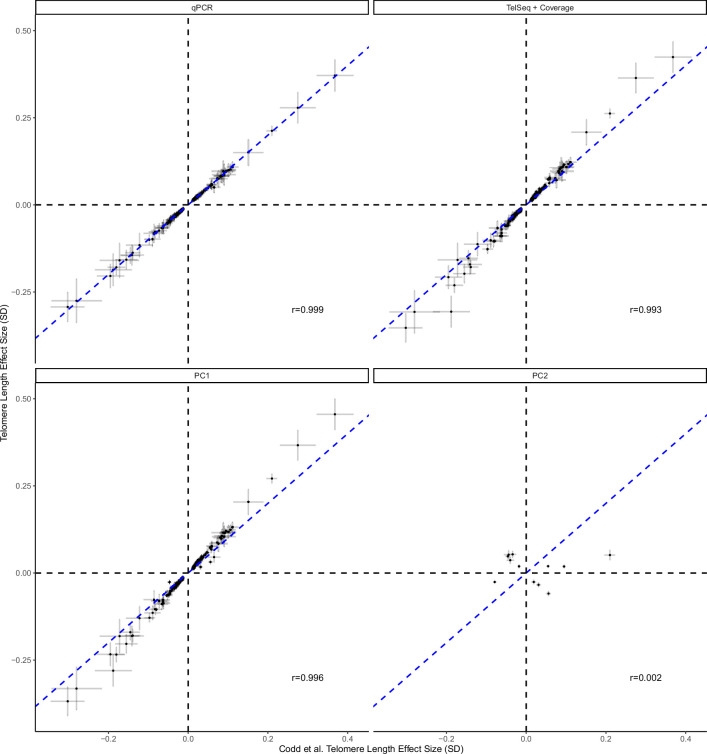
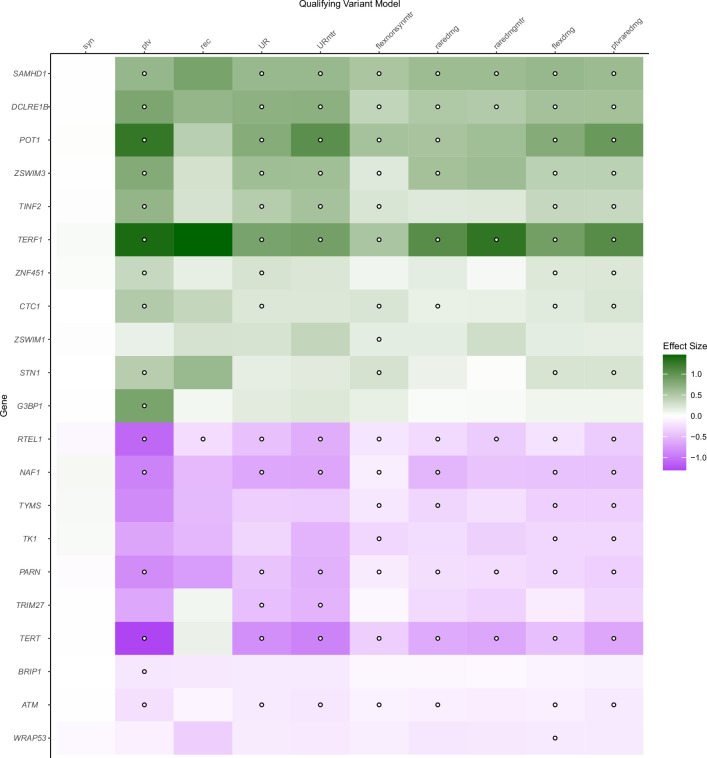
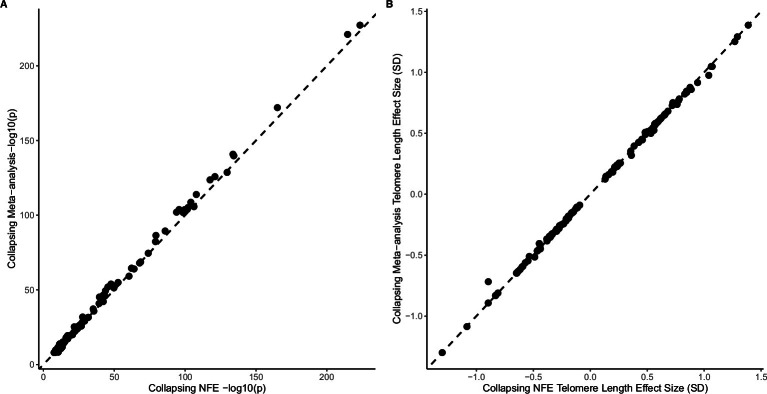
Telomeres protect chromosome ends from damage and their length is linked with human disease and aging. We developed a joint telomere length metric, combining quantitative PCR and whole-genome sequencing measurements from 462,666 UK Biobank participants. This metric increased SNP heritability, suggesting that it better captures genetic regulation of telomere length. Exome-wide rare-variant and gene-level collapsing association studies identified 64 variants and 30 genes significantly associated with telomere length, including allelic series in ACD and RTEL1. Notably, 16% of these genes are known drivers of clonal hematopoiesis-an age-related somatic mosaicism associated with myeloid cancers and several nonmalignant diseases. Somatic variant analyses revealed gene-specific associations with telomere length, including lengthened telomeres in individuals with large SRSF2-mutant clones, compared with shortened telomeres in individuals with clonal expansions driven by other genes. Collectively, our findings demonstrate the impact of rare variants on telomere length, with larger effects observed among genes also associated with clonal hematopoiesis.
© 2024. The Author(s).
Conflict of interest statement
O.S.B., R.S.D., S.V.V.D., S. Wen, A.N., J.M., F.H., D.S.L., K.R.S., N.R., H.O., A.P., P.V., Q. Wu, R.E.M., S. Wasilewski, K.C. M.F., Q. Wang, M.N.P. and S.P. are current employees and/or stockholders of AstraZeneca. All other authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
