

Comprehensive Overview of Bottom-Up Proteomics Using Mass Spectrometry
- PMID: 39193565
- PMCID: PMC11348894
- DOI: 10.1021/acsmeasuresciau.3c00068
Comprehensive Overview of Bottom-Up Proteomics Using Mass Spectrometry
Abstract
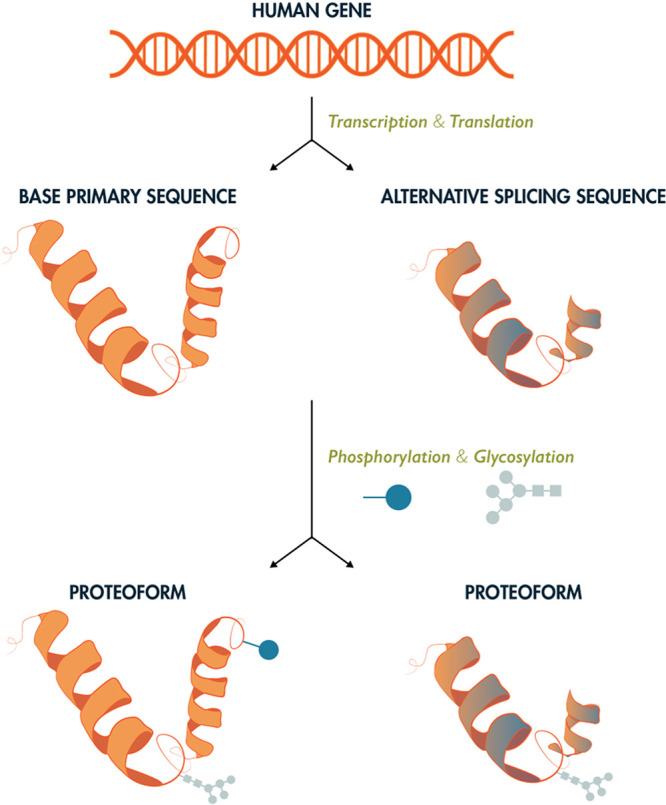
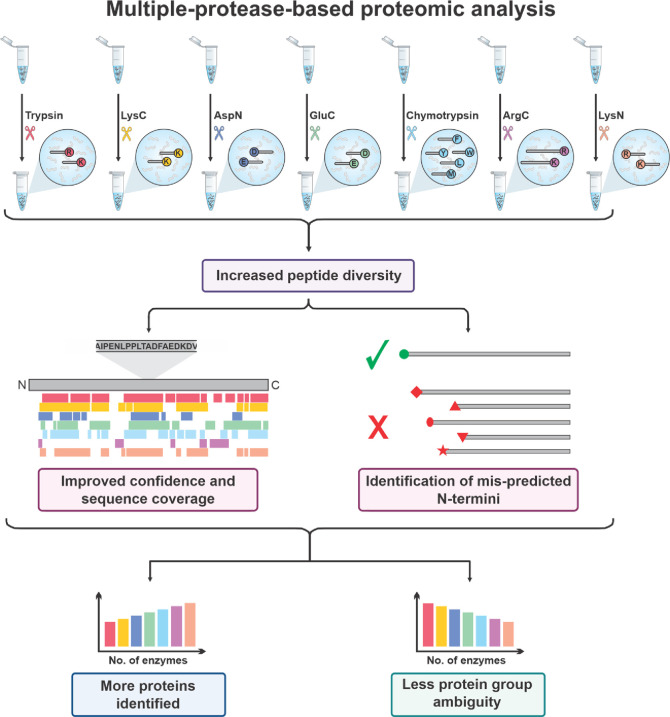
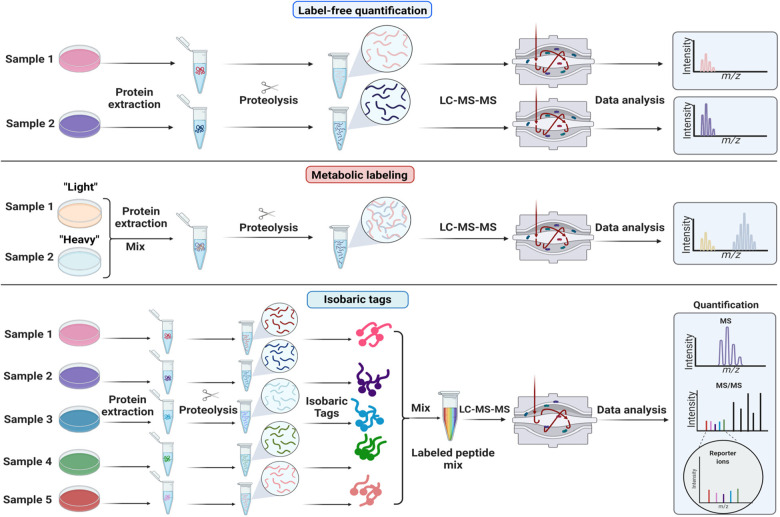
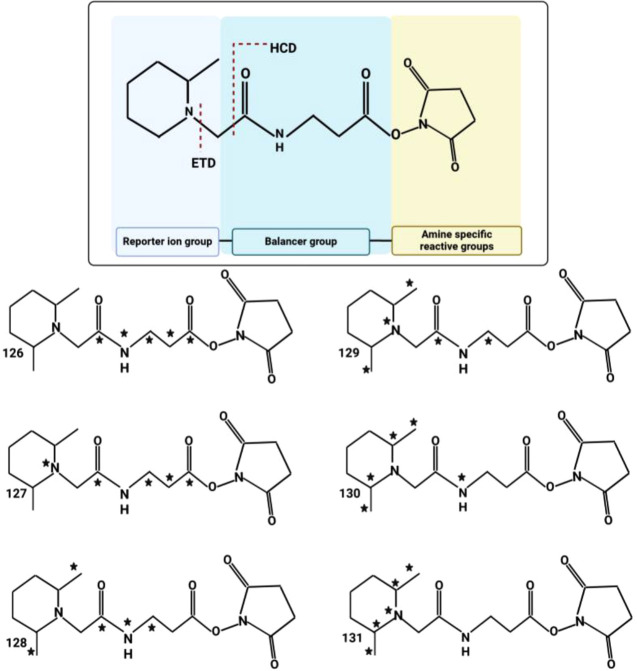
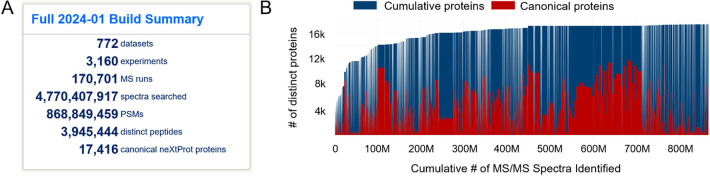
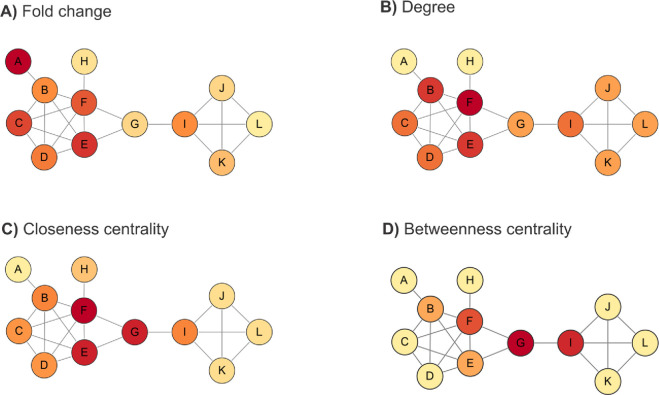
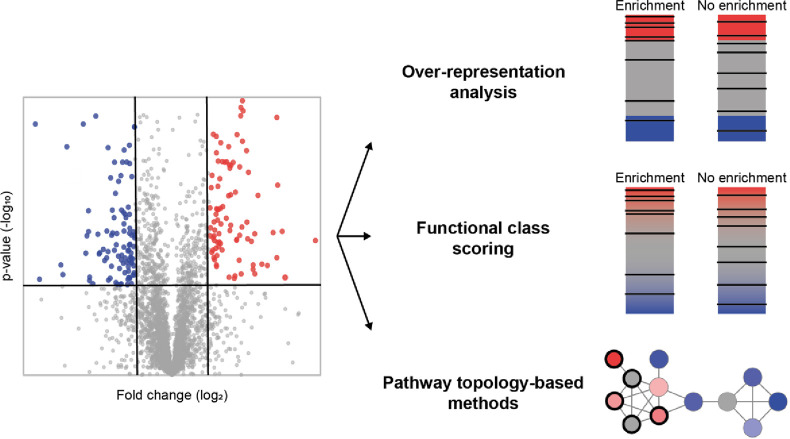
Proteomics is the large scale study of protein structure and function from biological systems through protein identification and quantification. "Shotgun proteomics" or "bottom-up proteomics" is the prevailing strategy, in which proteins are hydrolyzed into peptides that are analyzed by mass spectrometry. Proteomics studies can be applied to diverse studies ranging from simple protein identification to studies of proteoforms, protein-protein interactions, protein structural alterations, absolute and relative protein quantification, post-translational modifications, and protein stability. To enable this range of different experiments, there are diverse strategies for proteome analysis. The nuances of how proteomic workflows differ may be challenging to understand for new practitioners. Here, we provide a comprehensive overview of different proteomics methods. We cover from biochemistry basics and protein extraction to biological interpretation and orthogonal validation. We expect this Review will serve as a handbook for researchers who are new to the field of bottom-up proteomics.
© 2024 The Authors. Published by American Chemical Society.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
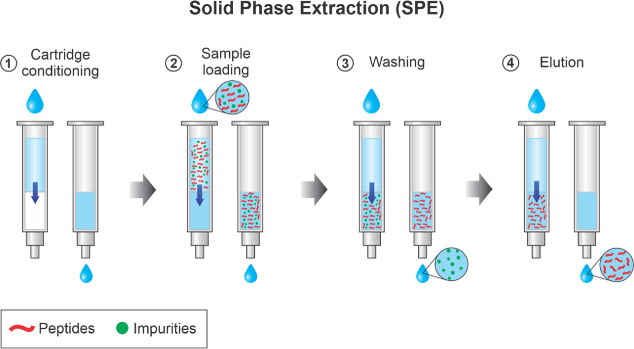
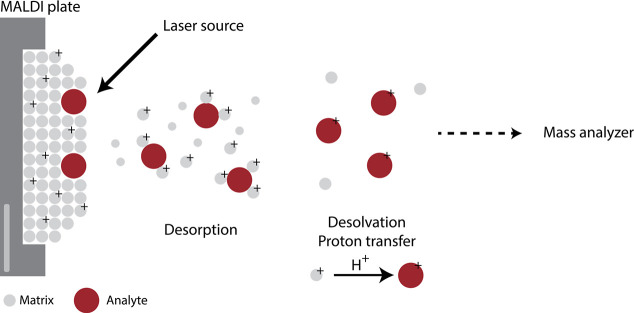
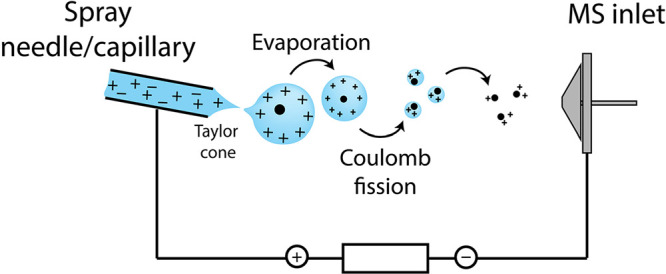
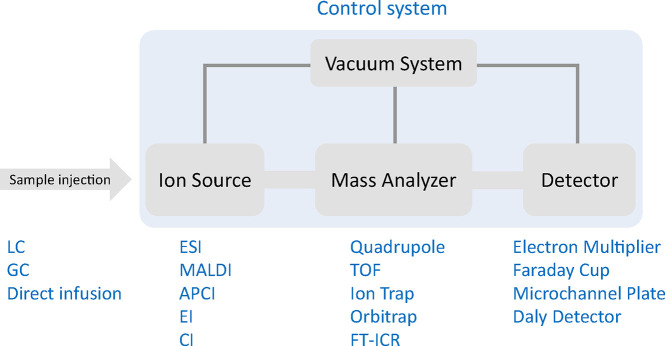
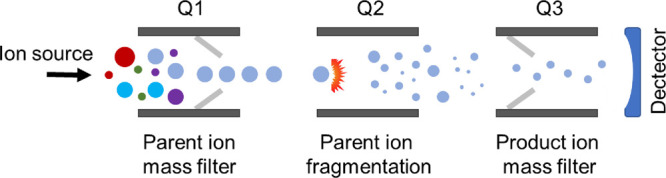
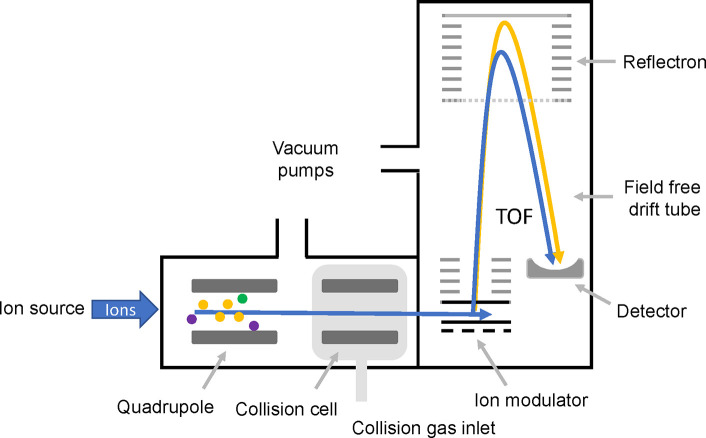
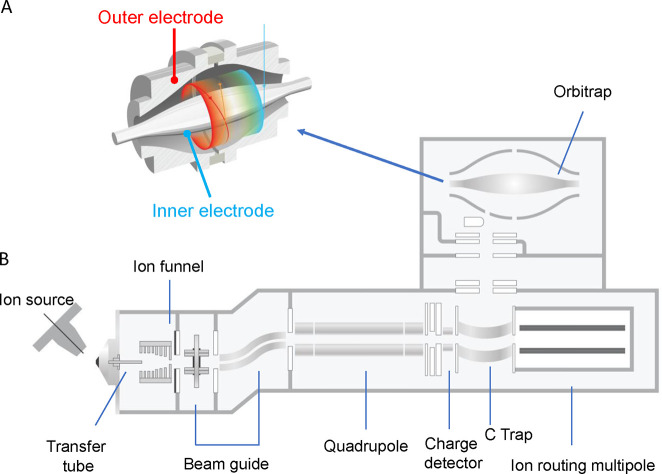
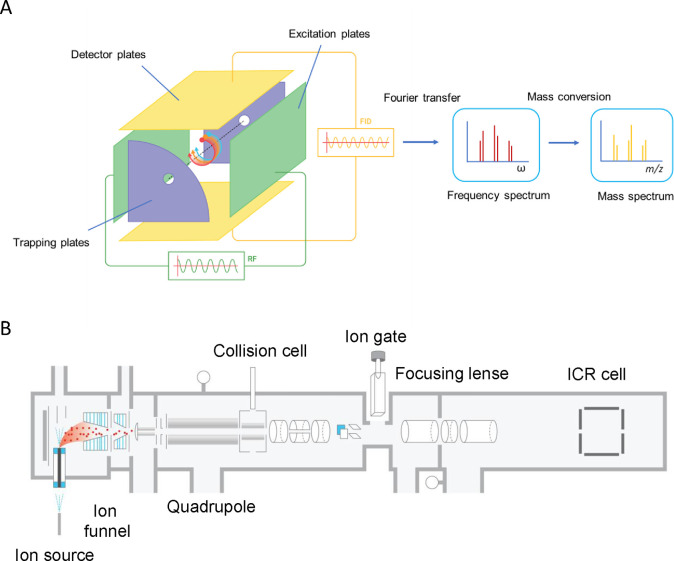
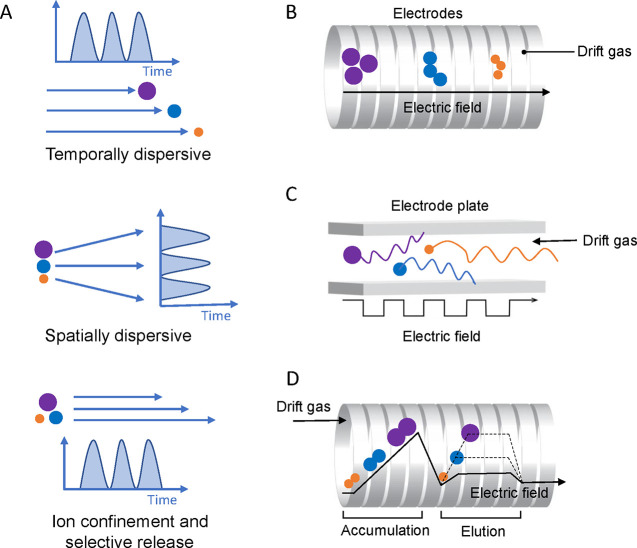
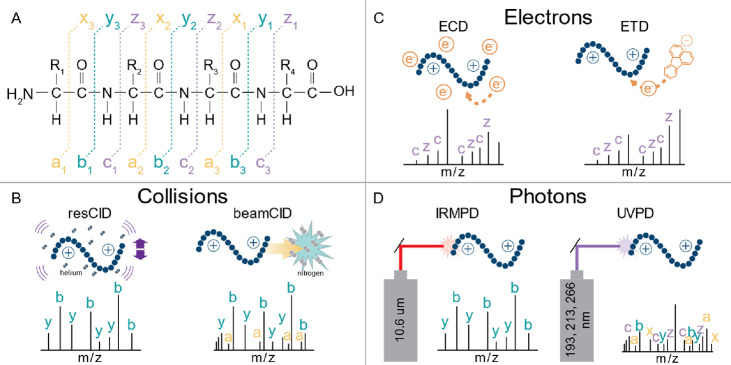
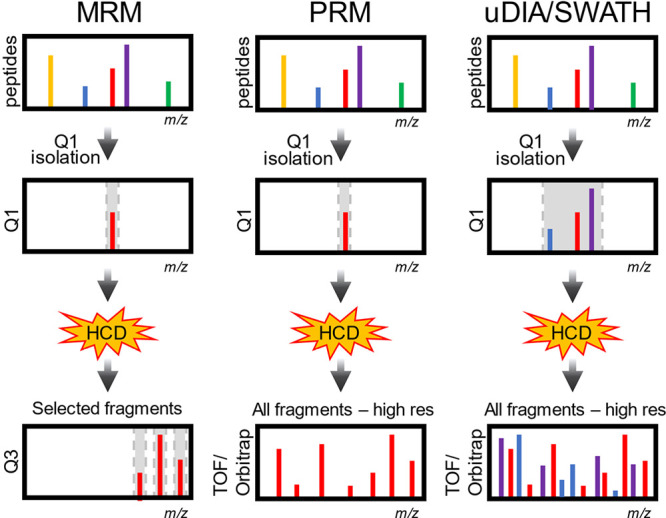
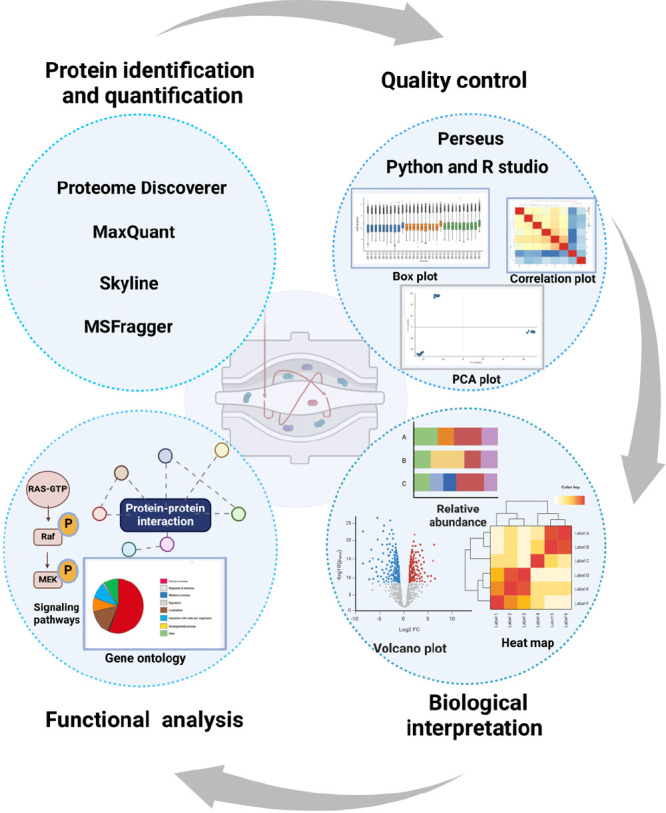
Update of
-
Comprehensive Overview of Bottom-Up Proteomics using Mass Spectrometry.ArXiv [Preprint]. 2023 Nov 13:arXiv:2311.07791v1. ArXiv. 2023. Update in: ACS Meas Sci Au. 2024 Jun 04;4(4):338-417. doi: 10.1021/acsmeasuresciau.3c00068. PMID: 38013887 Free PMC article. Updated. Preprint.
References
-
- Yusko E. C.; Bruhn B. R.; Eggenberger O. M.; Houghtaling J.; Rollings R. C.; Walsh N. C.; Nandivada S.; Pindrus M.; Hall A. R.; Sept D.; Li J.; Kalonia D. S.; Mayer M. Real-Time Shape Approximation and Fingerprinting of Single Proteins Using a Nanopore. Nature Nanotech 2017, 12 (4), 360–367. 10.1038/nnano.2016.267. - DOI - PubMed
-
- Swaminathan J.; Boulgakov A. A.; Hernandez E. T.; Bardo A. M.; Bachman J. L.; Marotta J.; Johnson A. M.; Anslyn E. V.; Marcotte E. M. Highly Parallel Single-Molecule Identification of Proteins in Zeptomole-Scale Mixtures. Nat Biotechnol 2018, 36 (11), 1076–1082. 10.1038/nbt.4278. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources