

Extensive identification of genes involved in congenital and structural heart disorders and cardiomyopathy
- PMID: 39195995
- PMCID: PMC11358025
- DOI: 10.1038/s44161-022-00018-8
Extensive identification of genes involved in congenital and structural heart disorders and cardiomyopathy
Erratum in
-
Publisher Correction: Extensive identification of genes involved in congenital and structural heart disorders and cardiomyopathy.Nat Cardiovasc Res. 2022 May;1(5):529-531. doi: 10.1038/s44161-022-00072-2. Nat Cardiovasc Res. 2022. PMID: 40263883 Free PMC article. No abstract available.
Abstract
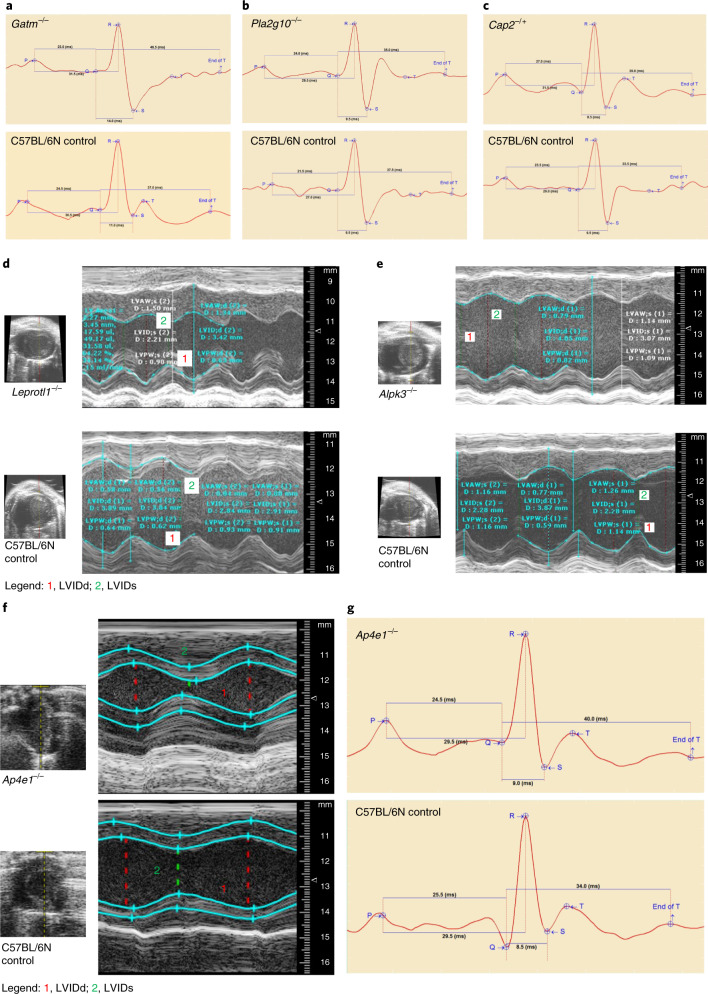
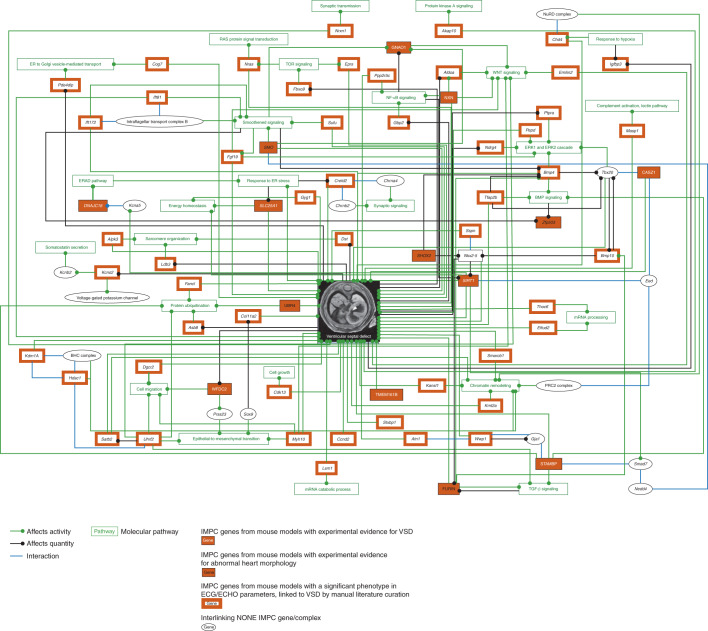
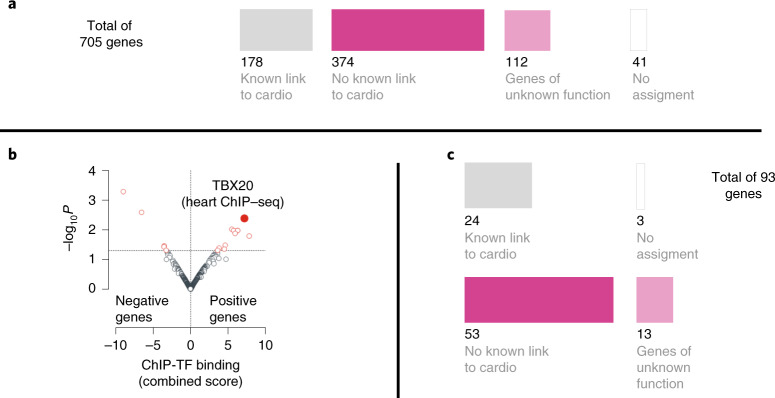
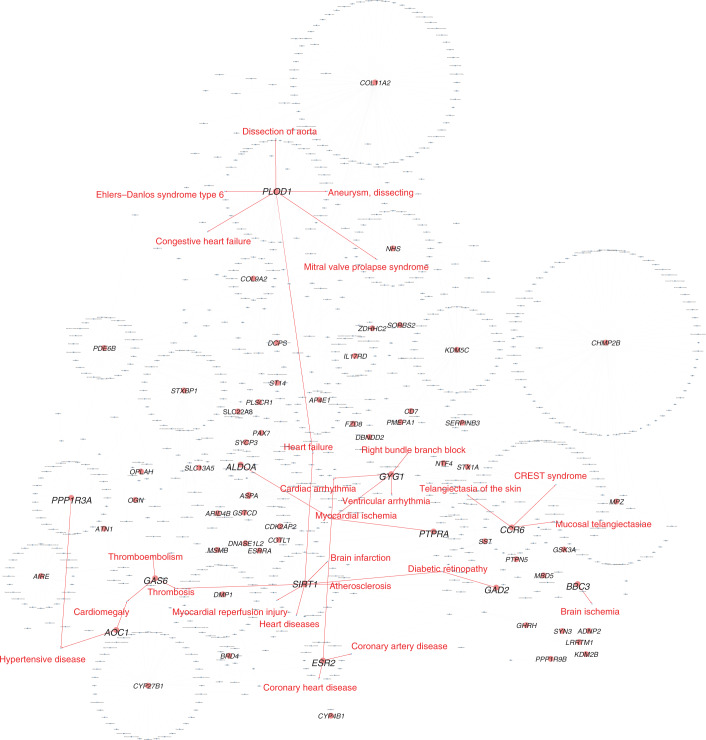
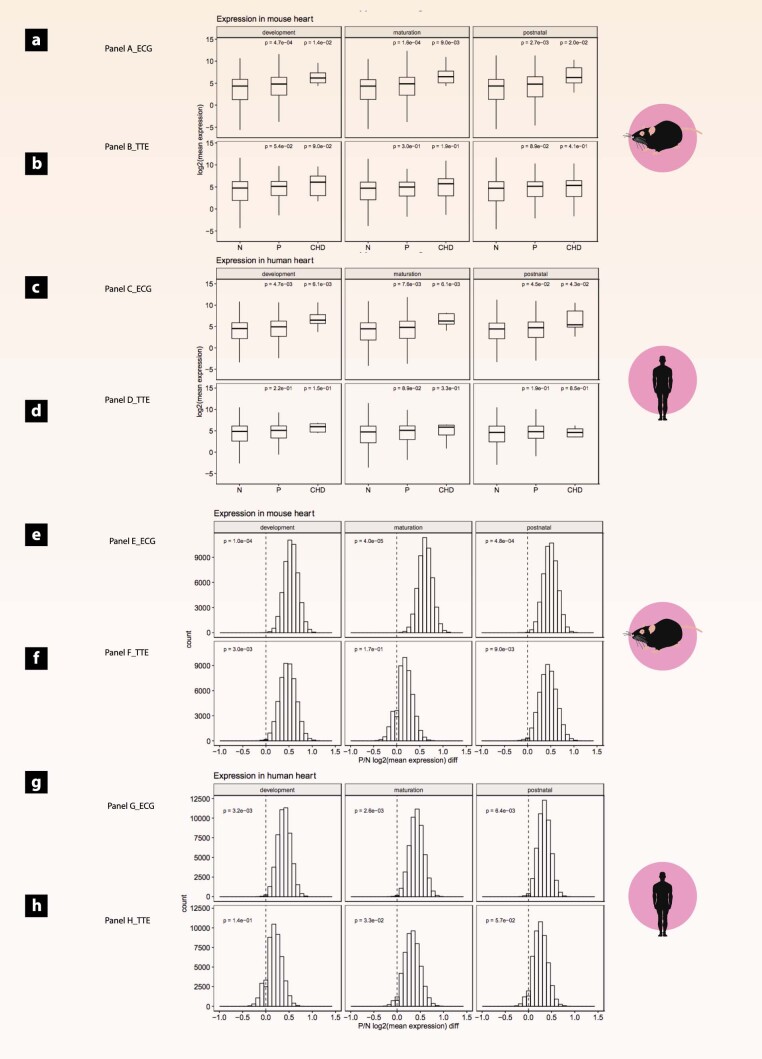
Clinical presentation of congenital heart disease is heterogeneous, making identification of the disease-causing genes and their genetic pathways and mechanisms of action challenging. By using in vivo electrocardiography, transthoracic echocardiography and microcomputed tomography imaging to screen 3,894 single-gene-null mouse lines for structural and functional cardiac abnormalities, here we identify 705 lines with cardiac arrhythmia, myocardial hypertrophy and/or ventricular dilation. Among these 705 genes, 486 have not been previously associated with cardiac dysfunction in humans, and some of them represent variants of unknown relevance (VUR). Mice with mutations in Casz1, Dnajc18, Pde4dip, Rnf38 or Tmem161b genes show developmental cardiac structural abnormalities, with their human orthologs being categorized as VUR. Using UK Biobank data, we validate the importance of the DNAJC18 gene for cardiac homeostasis by showing that its loss of function is associated with altered left ventricular systolic function. Our results identify hundreds of previously unappreciated genes with potential function in congenital heart disease and suggest causal function of five VUR in congenital heart disease.
© 2022. The Author(s).
Conflict of interest statement
T.I.O. has received honoraria or consulted for Abbott, AstraZeneca, Chiron, Genentech, Infinity Pharmaceuticals, Merz Pharmaceuticals, Merck Darmstadt, Mitsubishi Tanabe, Novartis, Ono Pharmaceuticals, Pfizer, Roche, Sanofi and Wyeth. None of these funds were used for this research project.
Figures


References
-
- Dakkak, W. & Oliver, T. I. Ventricular septal defect. in StatPearlshttps://www.ncbi.nlm.nih.gov/books/NBK470330/ (StatPearls, 2019). - PubMed
Grants and funding
- U01 HL098163/HL/NHLBI NIH HHS/United States
- U01 HL098188/HL/NHLBI NIH HHS/United States
- U01 HL098162/HL/NHLBI NIH HHS/United States
- MR/M009203/1/MRC_/Medical Research Council/United Kingdom
- UM1 OD023221/OD/NIH HHS/United States
- MC_EX_MR/M009203/1/MRC_/Medical Research Council/United Kingdom
- UM1 HG006370/HG/NHGRI NIH HHS/United States
- U01 HL098123/HL/NHLBI NIH HHS/United States
- U42 OD011175/OD/NIH HHS/United States
- U01 HL098147/HL/NHLBI NIH HHS/United States
- MC_UP_1502/1/MRC_/Medical Research Council/United Kingdom
- UM1 OD023222/OD/NIH HHS/United States
- U01 HL098153/HL/NHLBI NIH HHS/United States
- MC_PC_14089/MRC_/Medical Research Council/United Kingdom
- U54 HG006364/HG/NHGRI NIH HHS/United States
- UM1 HG006348/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases