

Cell-intrinsic effects of clonal hematopoiesis in heart failure
- PMID: 39196061
- PMCID: PMC11357996
- DOI: 10.1038/s44161-023-00322-x
Cell-intrinsic effects of clonal hematopoiesis in heart failure
Abstract
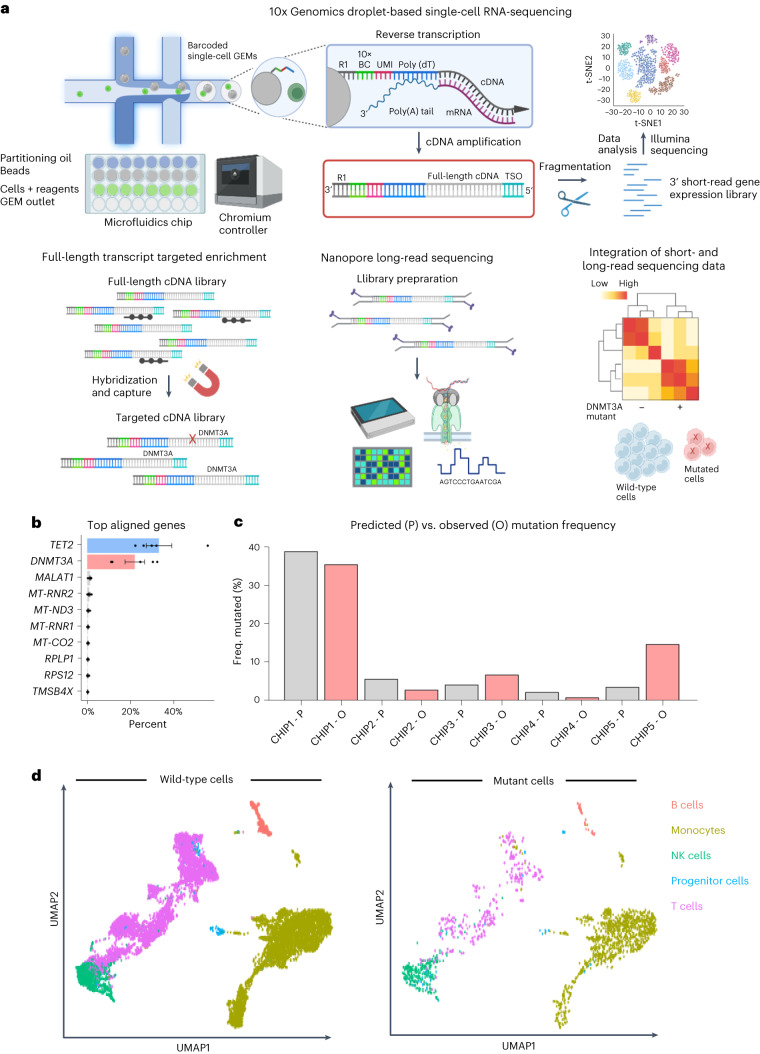
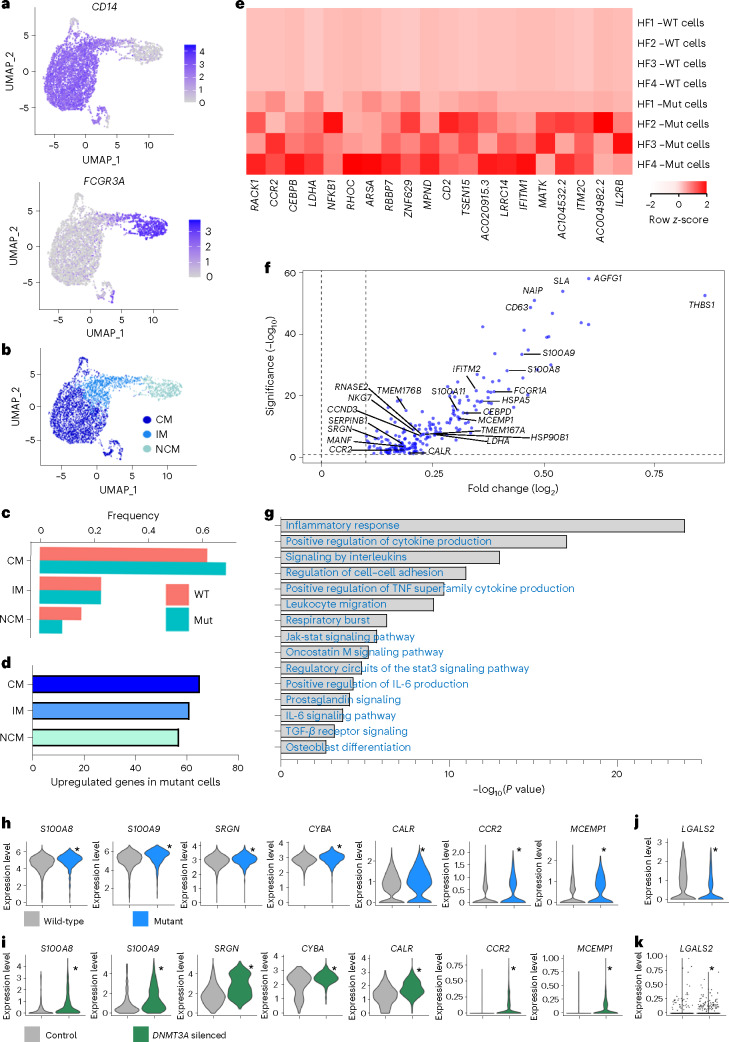
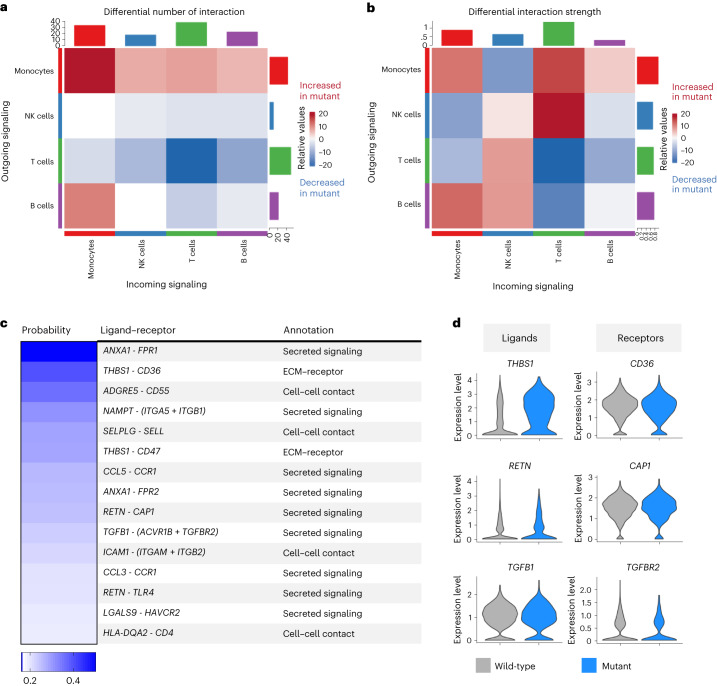
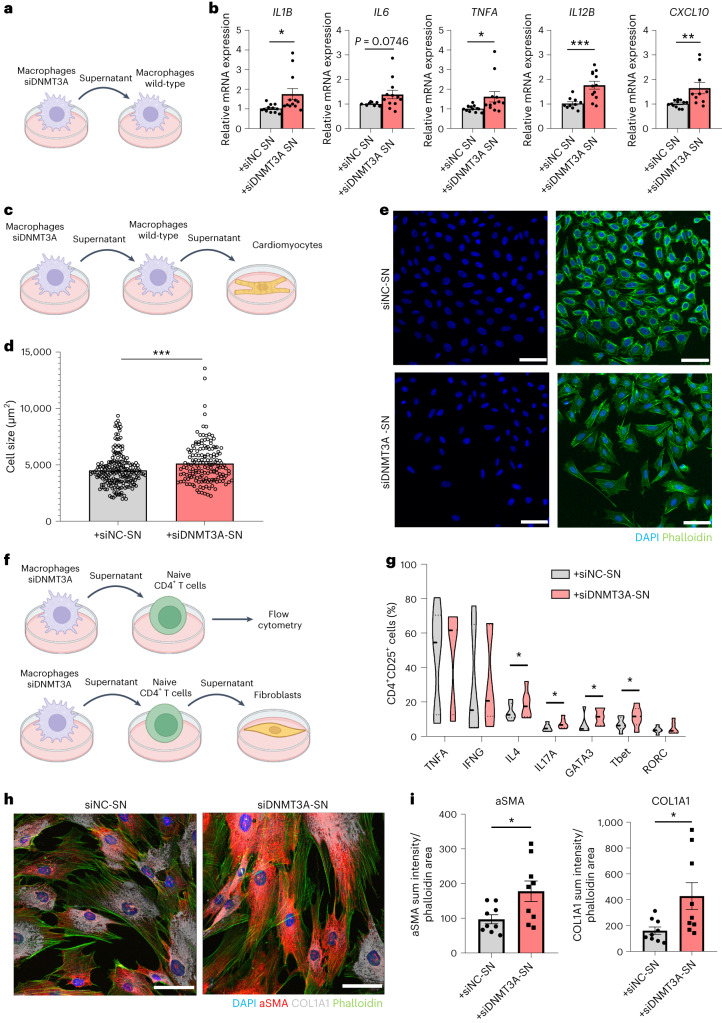
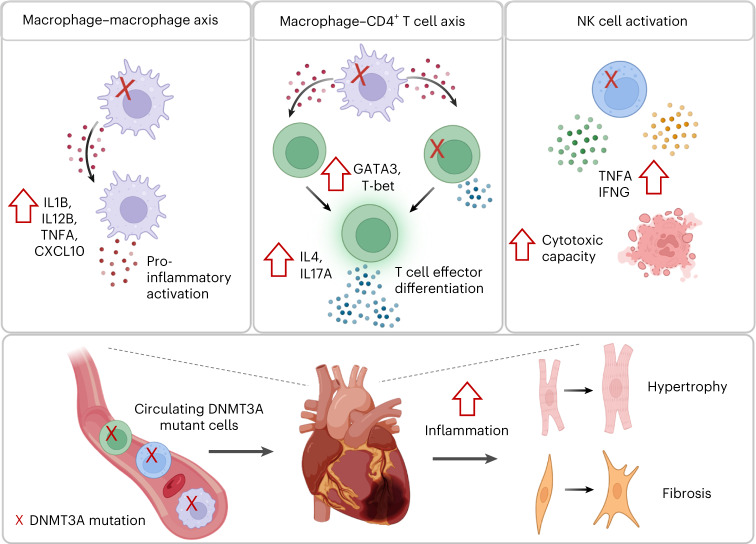
Clonal hematopoiesis of indeterminate potential (CHIP) is caused by somatic mutations in hematopoietic stem cells and associates with worse prognosis in patients with heart failure. Patients harboring CHIP mutations show enhanced inflammation. However, whether these signatures are derived from the relatively low number of cells harboring mutations or are indicators of systemic pro-inflammatory activation that is associated with CHIP is unclear. Here we assess the cell-intrinsic effects of CHIP mutant cells in patients with heart failure. Using an improved single-cell sequencing pipeline (MutDetect-Seq), we show that DNMT3A mutant monocytes, CD4+ T cells and NK cells exhibit altered gene expression profiles. While monocytes showed increased genes associated with inflammation and phagocytosis, T cells and NK cells present increased activation signatures and effector functions. Increased paracrine signaling pathways are predicted and validated between mutant and wild-type monocytes and T cells, which amplify inflammatory circuits. Altogether, these data provide novel insights into how CHIP might promote a worse prognosis in patients with heart failure.
© 2023. The Author(s).
Conflict of interest statement
A.M.Z. is an unpaid consultant for TenSixteen Bio. All remaining authors declare no competing interests.
Figures










References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials