

Plakophilin 2 gene therapy prevents and rescues arrhythmogenic right ventricular cardiomyopathy in a mouse model harboring patient genetics
- PMID: 39196150
- PMCID: PMC11357983
- DOI: 10.1038/s44161-023-00370-3
Plakophilin 2 gene therapy prevents and rescues arrhythmogenic right ventricular cardiomyopathy in a mouse model harboring patient genetics
Abstract
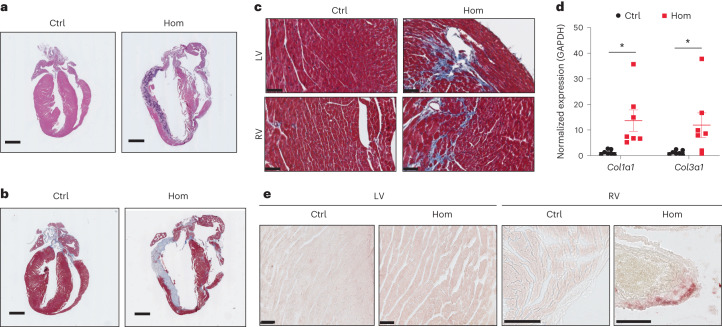
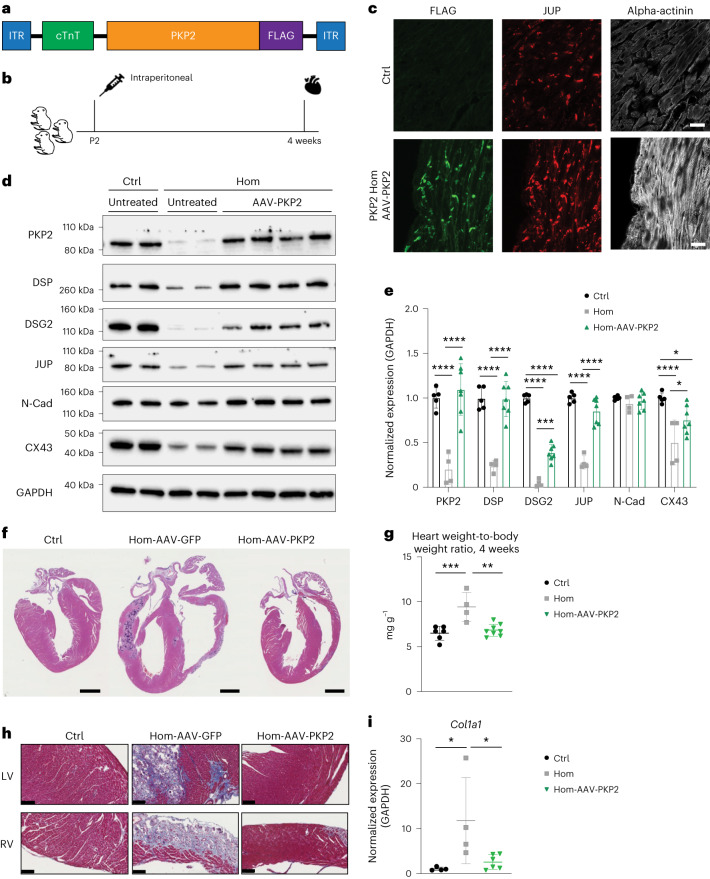
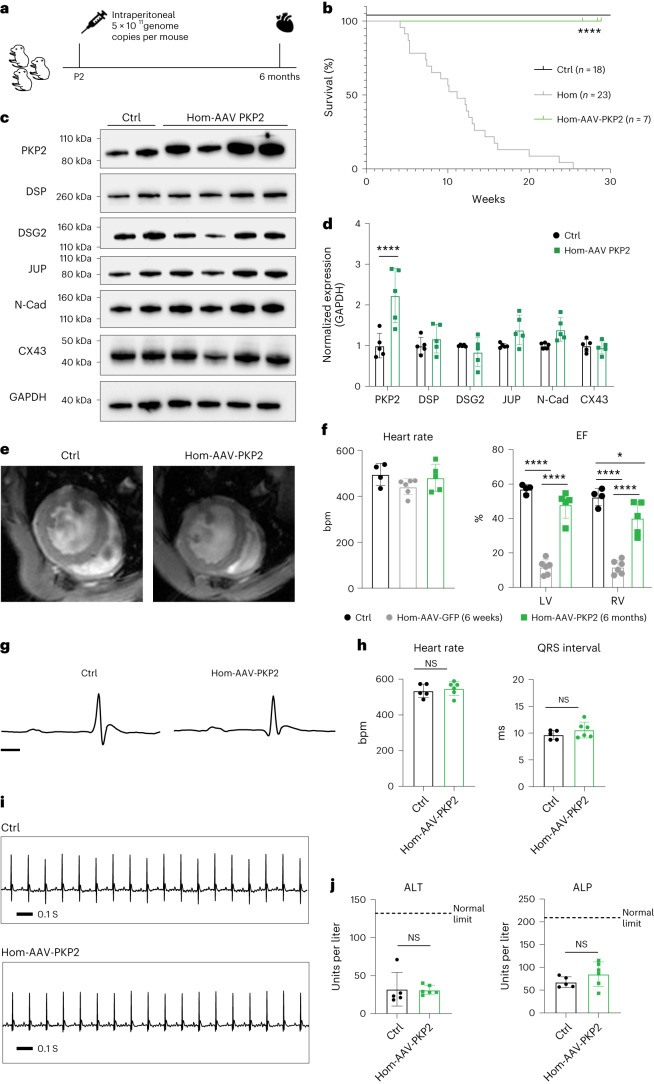
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a fatal genetic heart disease characterized by cardiac arrhythmias, in which fibrofatty deposition leads to heart failure, with no effective treatments. Plakophilin 2 (PKP2) is the most frequently mutated gene in ARVC, and although altered RNA splicing has been implicated, there are no models to study its effect and therapeutics. Here, we generate a mouse model harboring a PKP2 mutation (IVS10-1G>C) affecting RNA splicing, recapitulating ARVC features and sudden death starting at 4 weeks. Administering AAV-PKP2 gene therapy (adeno-associated viral therapy to drive cardiac expression of PKP2) to neonatal mice restored PKP2 protein levels, completely preventing cardiac desmosomal and pathological deficits associated with ARVC, ensuring 100% survival of mice up to 6 months. Late-stage AAV-PKP2 administration rescued desmosomal protein deficits and reduced pathological deficits including improved cardiac function in adult mice, resulting in 100% survival up to 4 months. We suggest that AAV-PKP2 gene therapy holds promise for circumventing ARVC associated with PKP2 mutations, including splice site mutations.
© 2023. The Author(s).
Conflict of interest statement
F.S. was a co-founder of Stelios Therapeutics (acquired by LEXEO Therapeutics) and is a co-founder and shareholder of Papillon Therapeutics Inc as well as a consultant and shareholder of LEXEO Therapeutics Inc. A.M. is a co-founder of and equity holder in Insilicomed Inc and Vektor Medical Inc. For A.M., findings reported in this study did not involve the companies he is involved with in any way, and these findings have no relationship with the business activities or scientific interests of either company. All other authors declare no competing interests.
Figures














Comment in
-
Restoring PKP2 in arrhythmogenic cardiomyopathy.Nat Cardiovasc Res. 2023 Dec;2(12):1115-1116. doi: 10.1038/s44161-023-00382-z. Nat Cardiovasc Res. 2023. PMID: 39196151 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL162369/HL/NHLBI NIH HHS/United States
- P30 NS047101/NS/NINDS NIH HHS/United States
- HL162369/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
- R01 HL121754/HL/NHLBI NIH HHS/United States
- HL121754/U.S. Department of Health & Human Services | NIH | National Heart, Lung, and Blood Institute (NHLBI)
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases