

A common gene signature of the right ventricle in failing rat and human hearts
- PMID: 39196177
- PMCID: PMC11358011
- DOI: 10.1038/s44161-024-00485-1
A common gene signature of the right ventricle in failing rat and human hearts
Abstract
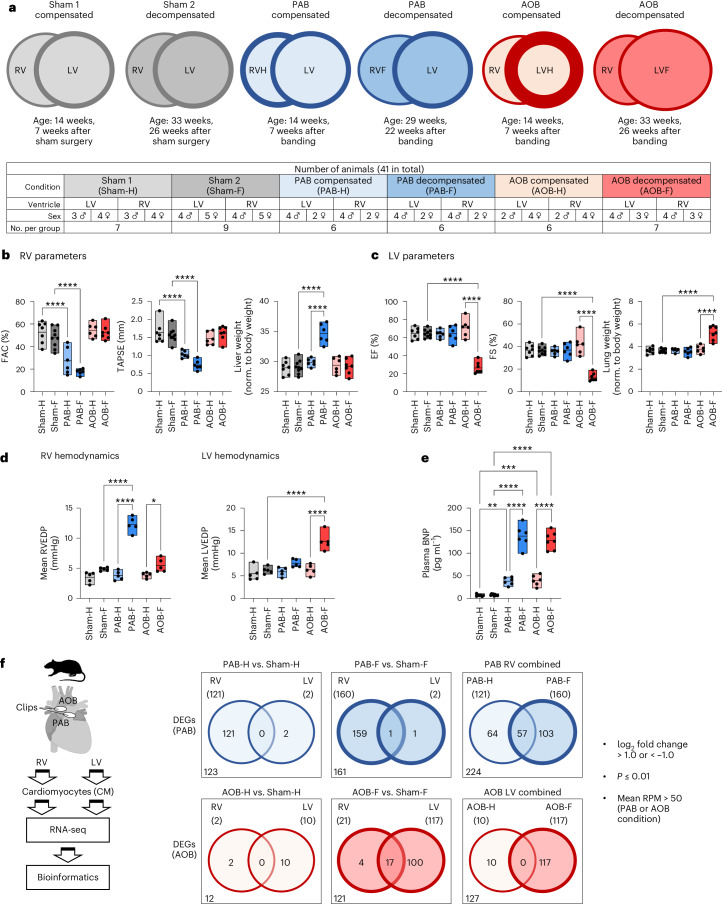
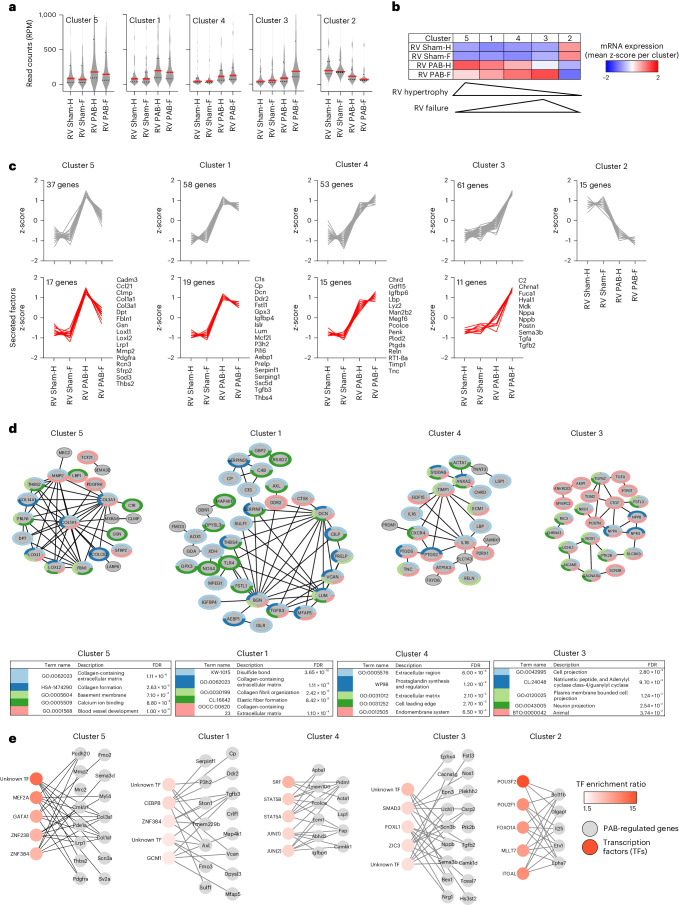
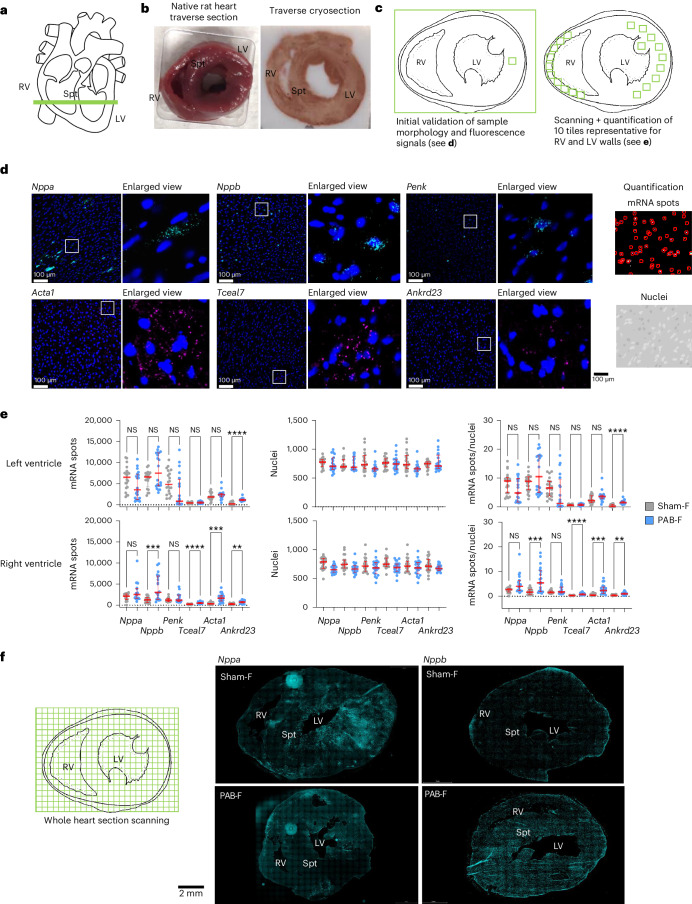
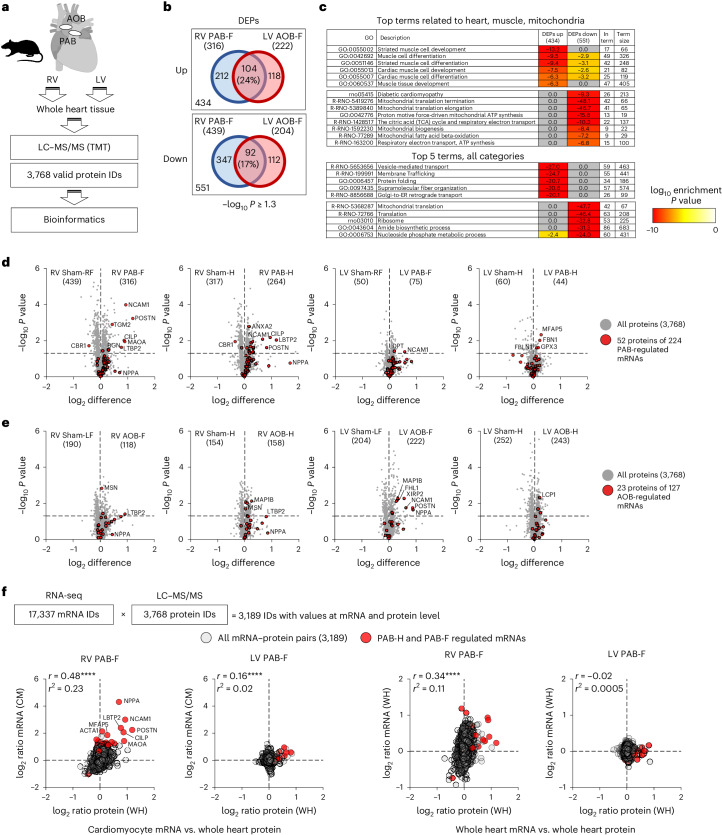
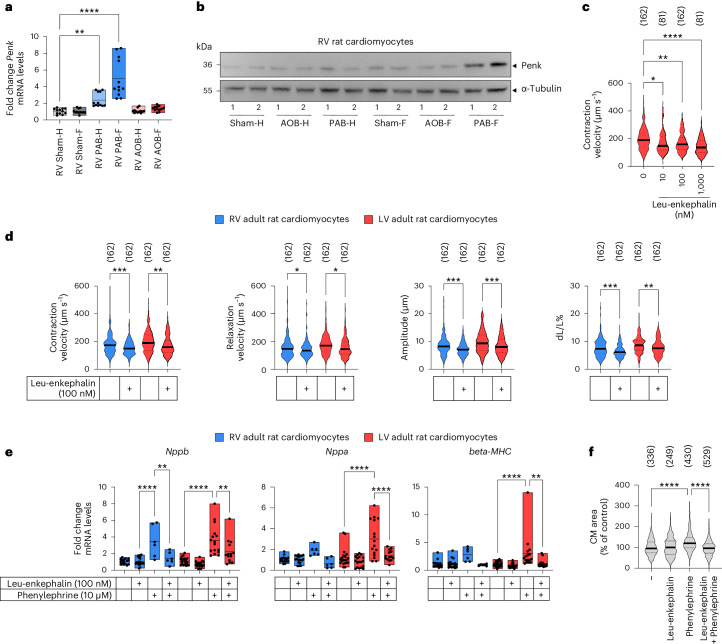
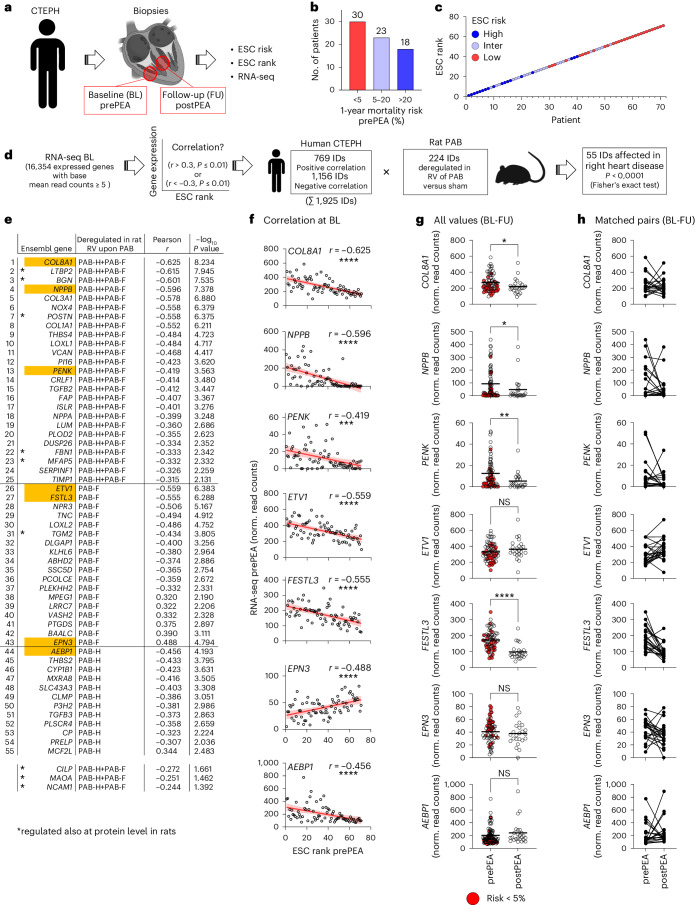
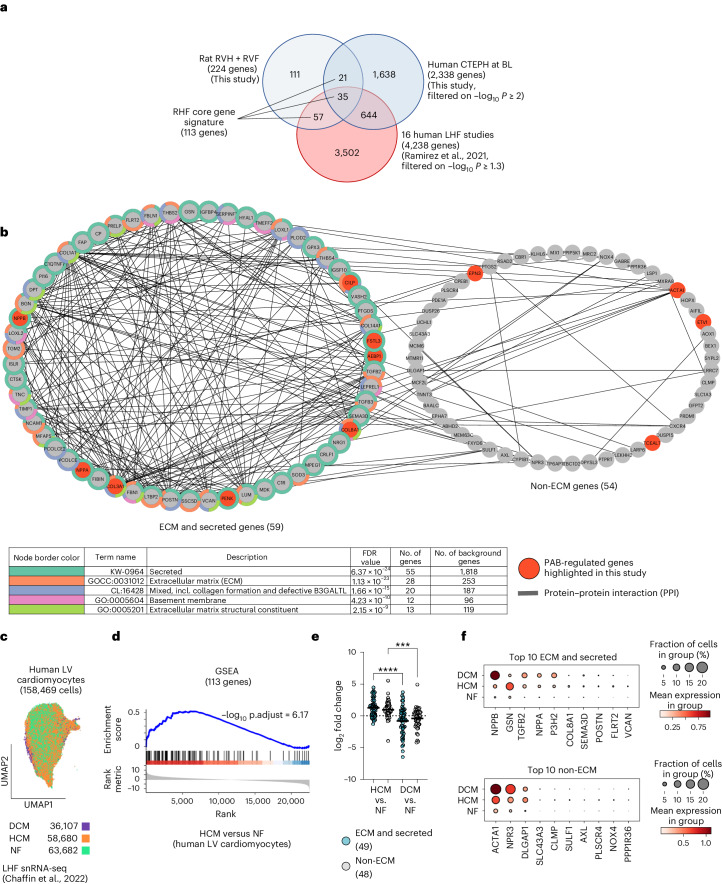
The molecular mechanisms of progressive right heart failure are incompletely understood. In this study, we systematically examined transcriptomic changes occurring over months in isolated cardiomyocytes or whole heart tissues from failing right and left ventricles in rat models of pulmonary artery banding (PAB) or aortic banding (AOB). Detailed bioinformatics analyses resulted in the identification of gene signature, protein and transcription factor networks specific to ventricles and compensated or decompensated disease states. Proteomic and RNA-FISH analyses confirmed PAB-mediated regulation of key genes and revealed spatially heterogeneous mRNA expression in the heart. Intersection of rat PAB-specific gene sets with transcriptome datasets from human patients with chronic thromboembolic pulmonary hypertension (CTEPH) led to the identification of more than 50 genes whose expression levels correlated with the severity of right heart disease, including multiple matrix-regulating and secreted factors. These data define a conserved, differentially regulated genetic network associated with right heart failure in rats and humans.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures











References
-
- Bozkurt, B. Contemporary pharmacological treatment and management of heart failure. Nat. Rev. Cardiol.10.1038/s41569-024-00997-0 (2024). - PubMed
MeSH terms
Grants and funding
- project 268555672/Deutsche Forschungsgemeinschaft (German Research Foundation)
- project 197785619/Deutsche Forschungsgemeinschaft (German Research Foundation)
- project 416910386/Deutsche Forschungsgemeinschaft (German Research Foundation)
- project 390649896/Deutsche Forschungsgemeinschaft (German Research Foundation)
- project 268555672/Deutsche Forschungsgemeinschaft (German Research Foundation)
- project 268555672/Deutsche Forschungsgemeinschaft (German Research Foundation)
- project 268555672/Deutsche Forschungsgemeinschaft (German Research Foundation)
- project 268555672/Deutsche Forschungsgemeinschaft (German Research Foundation)
- project 268555672/Deutsche Forschungsgemeinschaft (German Research Foundation)
- project 268555672/Deutsche Forschungsgemeinschaft (German Research Foundation)
- project 268555672/Deutsche Forschungsgemeinschaft (German Research Foundation)
- project 268555672/Deutsche Forschungsgemeinschaft (German Research Foundation)
- project 268555672/Deutsche Forschungsgemeinschaft (German Research Foundation)
LinkOut - more resources
Full Text Sources
Medical