

Cellular heterogeneity of pluripotent stem cell-derived cardiomyocyte grafts is mechanistically linked to treatable arrhythmias
- PMID: 39196193
- PMCID: PMC11358004
- DOI: 10.1038/s44161-023-00419-3
Cellular heterogeneity of pluripotent stem cell-derived cardiomyocyte grafts is mechanistically linked to treatable arrhythmias
Abstract
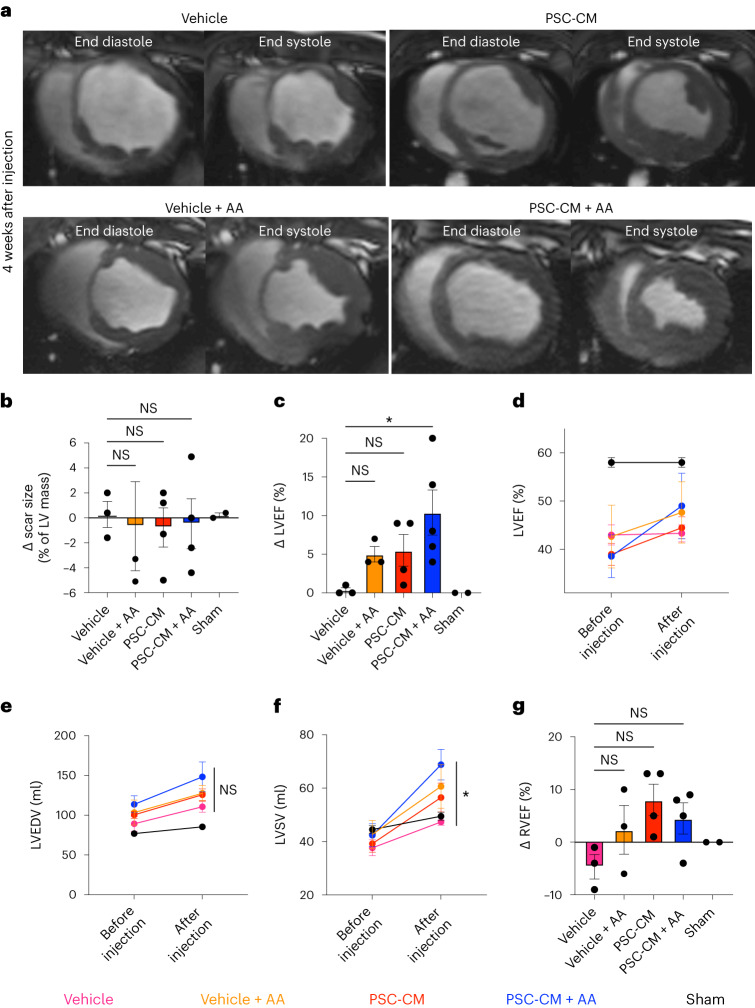
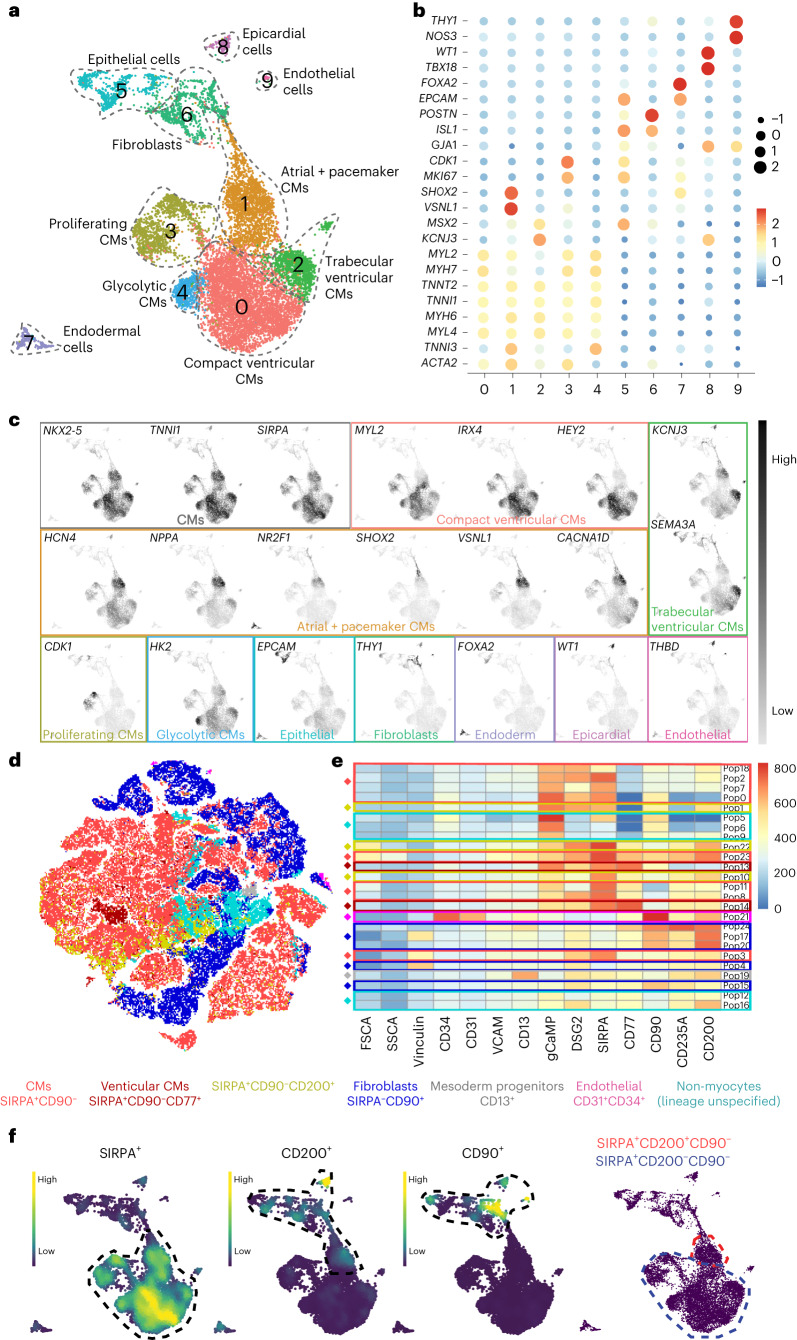
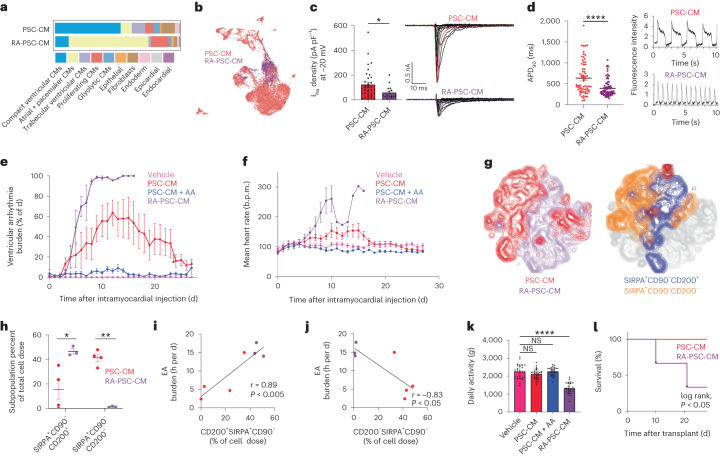
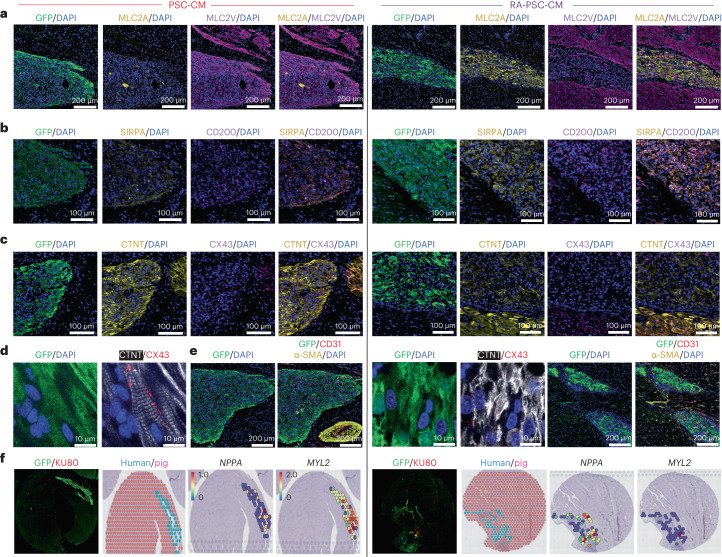
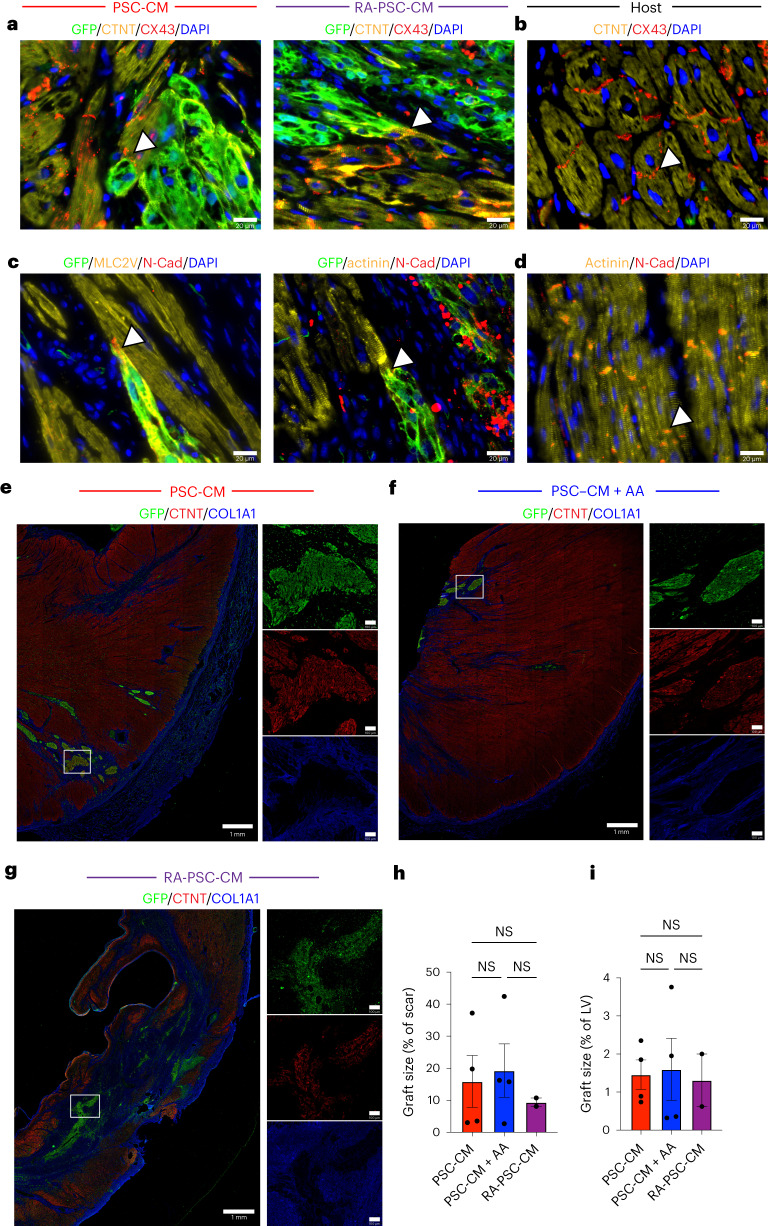
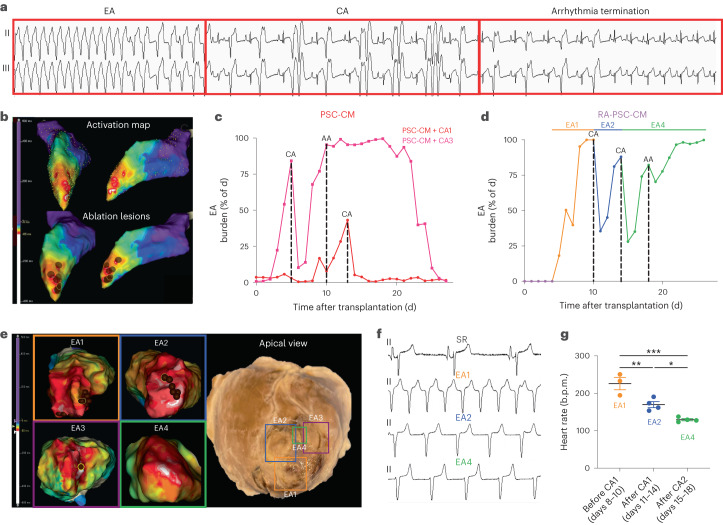
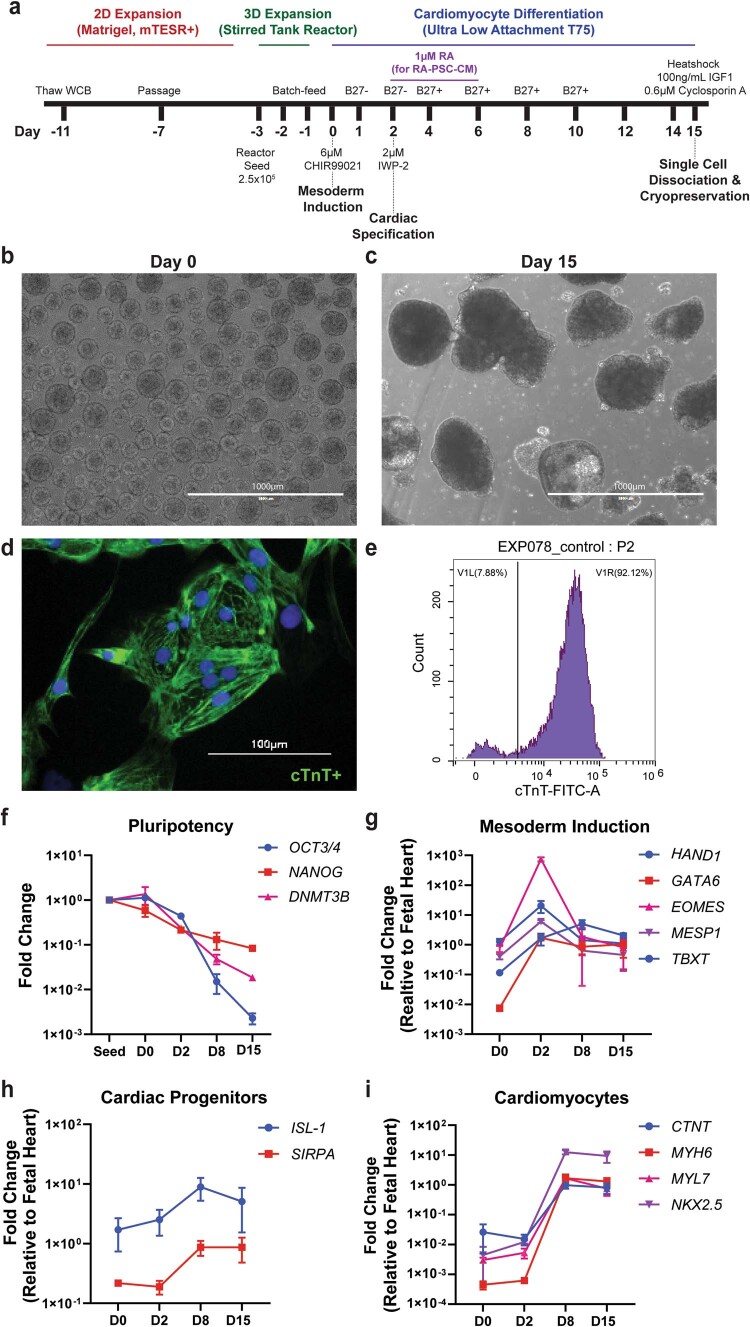
Preclinical data have confirmed that human pluripotent stem cell-derived cardiomyocytes (PSC-CMs) can remuscularize the injured or diseased heart, with several clinical trials now in planning or recruitment stages. However, because ventricular arrhythmias represent a complication following engraftment of intramyocardially injected PSC-CMs, it is necessary to provide treatment strategies to control or prevent engraftment arrhythmias (EAs). Here, we show in a porcine model of myocardial infarction and PSC-CM transplantation that EAs are mechanistically linked to cellular heterogeneity in the input PSC-CM and resultant graft. Specifically, we identify atrial and pacemaker-like cardiomyocytes as culprit arrhythmogenic subpopulations. Two unique surface marker signatures, signal regulatory protein α (SIRPA)+CD90-CD200+ and SIRPA+CD90-CD200-, identify arrhythmogenic and non-arrhythmogenic cardiomyocytes, respectively. Our data suggest that modifications to current PSC-CM-production and/or PSC-CM-selection protocols could potentially prevent EAs. We further show that pharmacologic and interventional anti-arrhythmic strategies can control and potentially abolish these arrhythmias.
© 2024. The Author(s).
Conflict of interest statement
J.J.H.C. reports financial compensation from Novo Nordisk and AstraZeneca related to educational activities and consultancy fees from Implicit Bioscience. All other authors declare no remaining competing interests.
Figures










References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical