

Hereditary C1q Deficiency is Associated with Type 1 Interferon-Pathway Activation and a High Risk of Central Nervous System Inflammation
- PMID: 39196411
- PMCID: PMC11358312
- DOI: 10.1007/s10875-024-01788-5
Hereditary C1q Deficiency is Associated with Type 1 Interferon-Pathway Activation and a High Risk of Central Nervous System Inflammation
Abstract
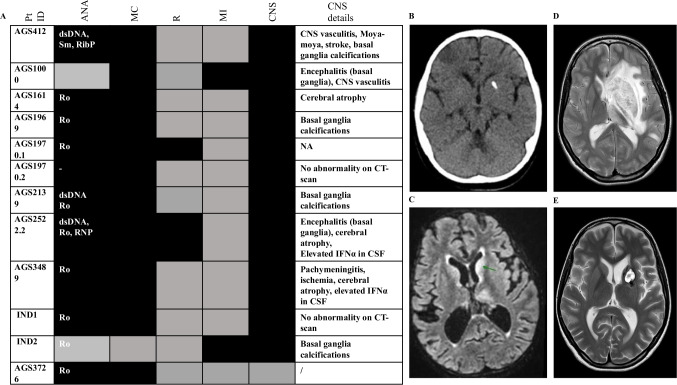
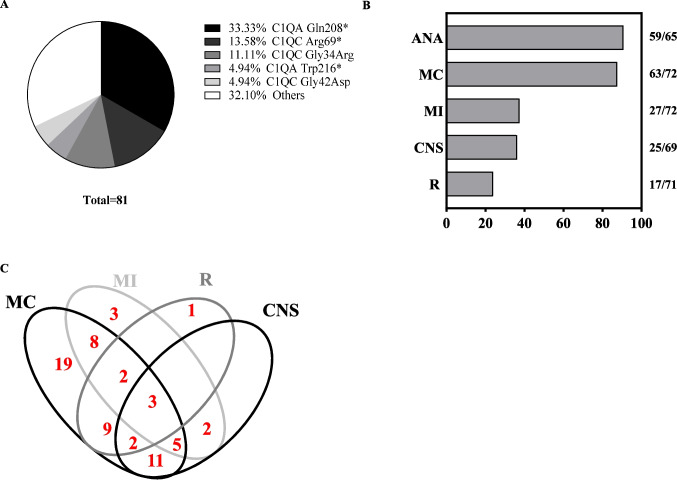
Hereditary C1q deficiency (C1QDef) is a rare monogenic disorder leading to defective complement pathway activation and systemic lupus erythematosus (SLE)-like manifestations. The link between impairment of the complement cascade and autoimmunity remains incompletely understood. Here, we assessed type 1 interferon pathway activation in patients with C1QDef. Twelve patients with genetically confirmed C1QDef were recruited through an international collaboration. Clinical, biological and radiological data were collected retrospectively. The expression of a standardized panel of interferon stimulated genes (ISGs) in peripheral blood was measured, and the level of interferon alpha (IFNα) protein in cerebrospinal fluid (CSF) determined using SIMOA technology. Central nervous system (encompassing basal ganglia calcification, encephalitis, vasculitis, chronic pachymeningitis), mucocutaneous and renal involvement were present, respectively, in 10, 11 and 2 of 12 patients, and severe infections recorded in 2/12 patients. Elevated ISG expression was observed in all patients tested (n = 10/10), and serum and CSF IFNα elevated in 2/2 patients. Three patients were treated with Janus-kinase inhibitors (JAKi), with variable outcome; one displaying an apparently favourable response in respect of cutaneous and neurological features, and two others experiencing persistent disease despite JAKi therapy. To our knowledge, we report the largest original series of genetically confirmed C1QDef yet described. Additionally, we present a review of all previously described genetically confirmed cases of C1QDef. Overall, individuals with C1QDef demonstrate many characteristics of recognized monogenic interferonopathies: particularly, cutaneous involvement (malar rash, acral vasculitic/papular rash, chilblains), SLE-like disease, basal ganglia calcification, increased expression of ISGs in peripheral blood, and elevated levels of CSF IFNα.
Keywords: C1Q deficiency; Complement; Janus-kinase inhibition; interferon; neuroinflammation; systemic lupus erythematosus.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
