

Recent evolutionary origin and localized diversity hotspots of mammalian coronaviruses
- PMID: 39196812
- PMCID: PMC11357359
- DOI: 10.7554/eLife.91745
Recent evolutionary origin and localized diversity hotspots of mammalian coronaviruses
Abstract
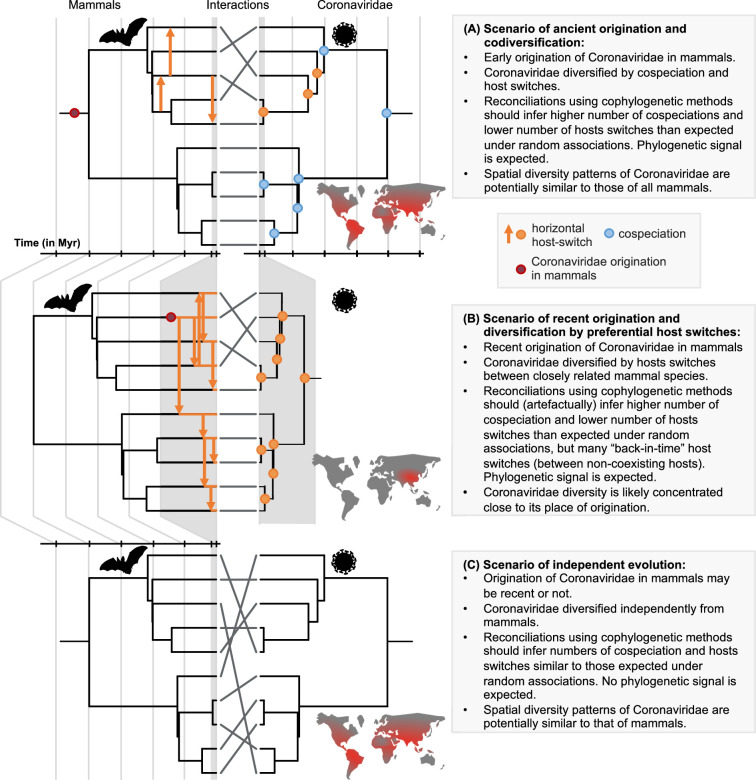
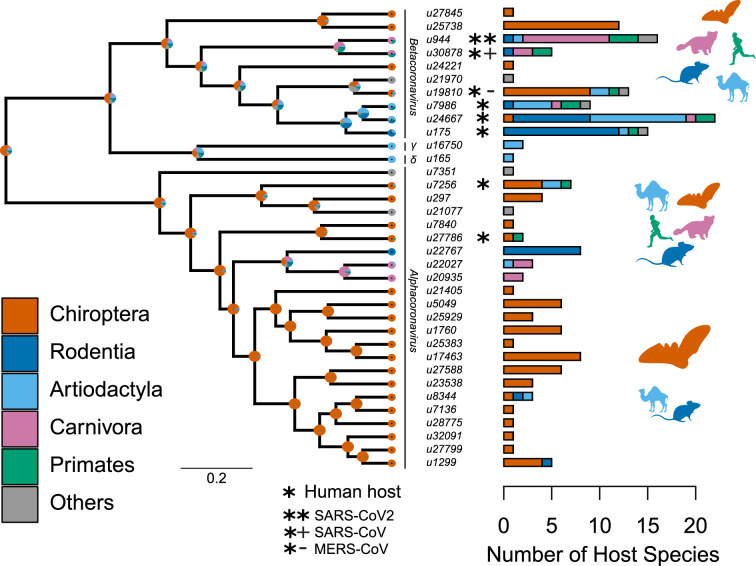
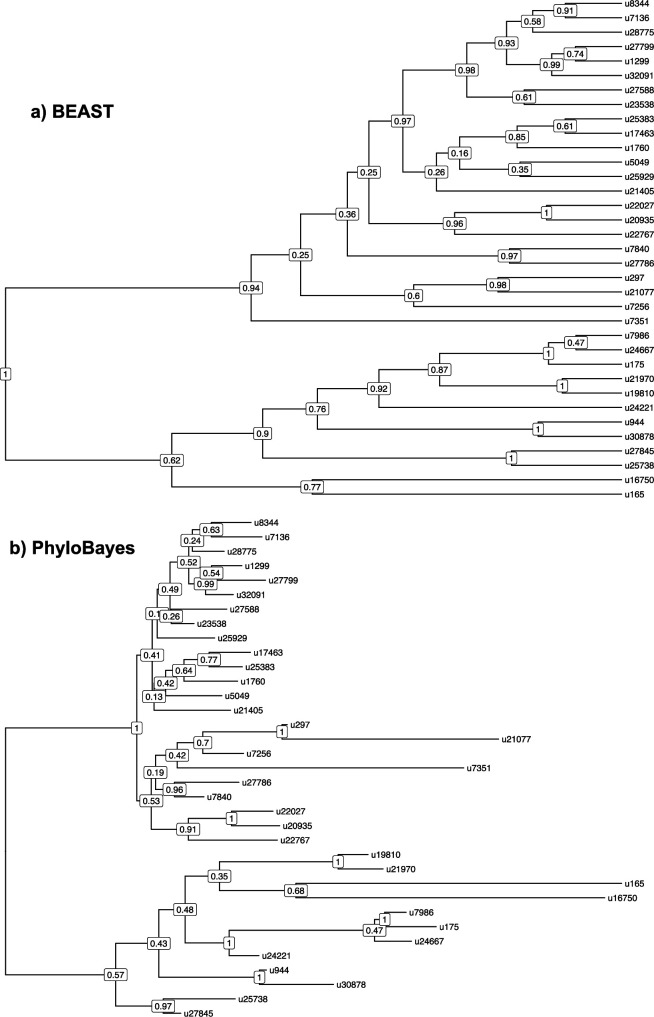
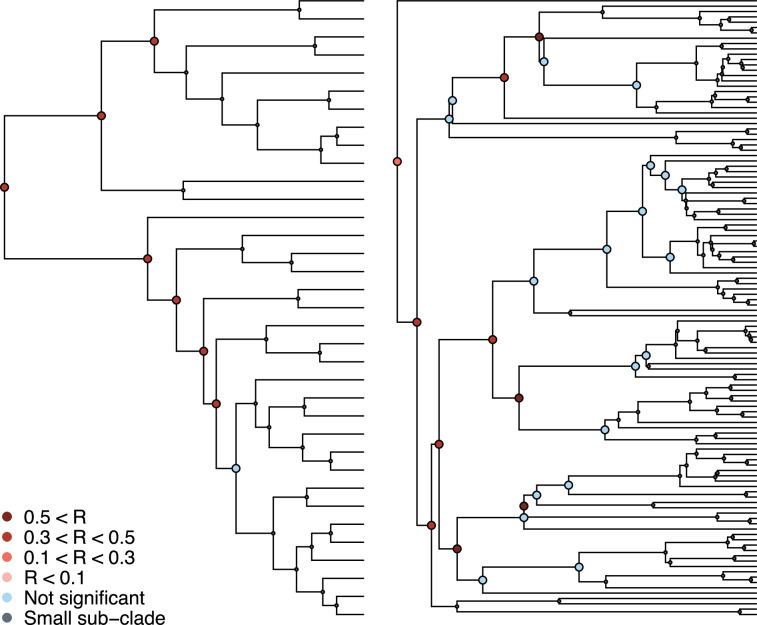
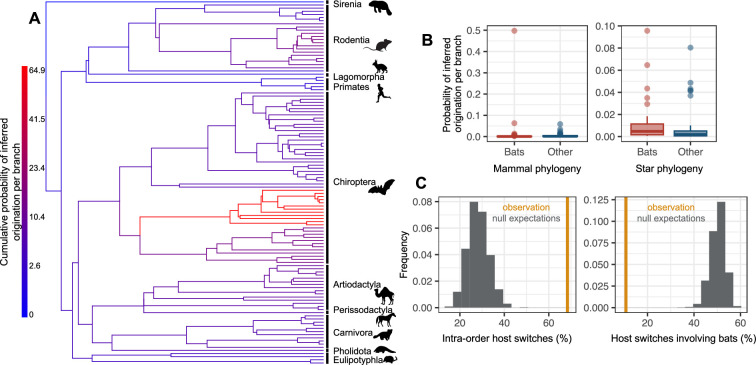
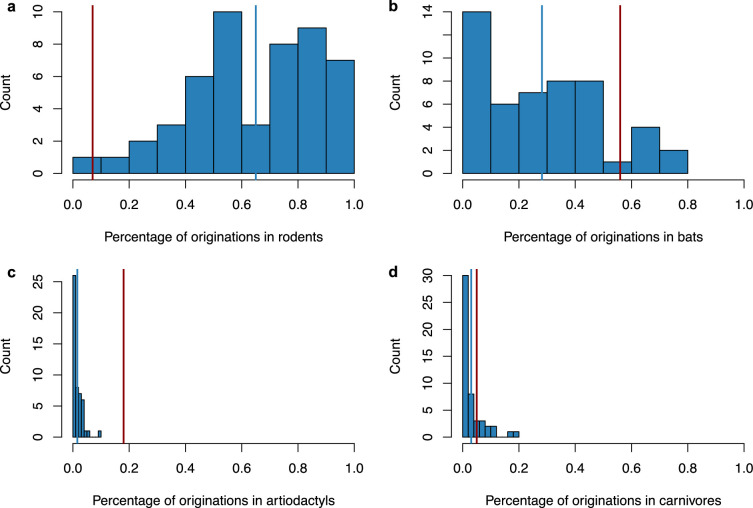
Several coronaviruses infect humans, with three, including the SARS-CoV2, causing diseases. While coronaviruses are especially prone to induce pandemics, we know little about their evolutionary history, host-to-host transmissions, and biogeography. One of the difficulties lies in dating the origination of the family, a particularly challenging task for RNA viruses in general. Previous cophylogenetic tests of virus-host associations, including in the Coronaviridae family, have suggested a virus-host codiversification history stretching many millions of years. Here, we establish a framework for robustly testing scenarios of ancient origination and codiversification versus recent origination and diversification by host switches. Applied to coronaviruses and their mammalian hosts, our results support a scenario of recent origination of coronaviruses in bats and diversification by host switches, with preferential host switches within mammalian orders. Hotspots of coronavirus diversity, concentrated in East Asia and Europe, are consistent with this scenario of relatively recent origination and localized host switches. Spillovers from bats to other species are rare, but have the highest probability to be towards humans than to any other mammal species, implicating humans as the evolutionary intermediate host. The high host-switching rates within orders, as well as between humans, domesticated mammals, and non-flying wild mammals, indicates the potential for rapid additional spreading of coronaviruses across the world. Our results suggest that the evolutionary history of extant mammalian coronaviruses is recent, and that cases of long-term virus-host codiversification have been largely over-estimated.
Keywords: codiversification; coevolution; coronavirus evolution; diversity of coronaviruses; epidemiology; evolutionary biology; global health; human; parasite diversification; preferential host switching; virus.
Plain language summary
The SARS-CoV-2 virus, which caused the recent global coronavirus pandemic, is the latest in a string of coronaviruses that have caused serious outbreaks. This group of coronaviruses can also infect other mammals and likely jumped between species – including from non-humans to humans – over the course of evolution. Determining when and how viruses evolved to infect humans can help scientists predict and prevent outbreaks. However, tracking the evolutionary trajectory of coronaviruses is challenging, and there are conflicting views on how often coronaviruses crossed between species and when these transitions likely occurred. Some studies suggest that coronaviruses originated early on in evolution and evolved together with their mammalian hosts, only occasionally jumping to and from different species. While others suggest they appeared more recently, and rapidly diversified by regularly transferring between species. To determine which is the most likely scenario, Maestri, Perez-Lamarque et al. developed a computational approach using already available data on the genetics and evolutionary history of mammals and coronaviruses. This revealed that coronaviruses originated recently in bats from East Asia and Europe, and primarily evolved by rapidly transferring between different mammalian species. This has led to geographical hotspots of diverse coronaviruses in East Asia and Europe. Maestri, Perez-Lamarque et al. found that it was rare for coronaviruses to spill over from bats to other types of mammals. Most of these spillovers resulted from coronaviruses jumping from bats to humans or domesticated animals. Humans appeared to be the main intermediary host that coronaviruses temporarily infected as they transferred from bats to other mammals. These findings – that coronaviruses emerged recently in evolution, jumped relatively frequently between species, and are geographically restricted – suggest that future transmissions are likely. Gathering more coronavirus samples from across the world and using even more powerful analysis tools could help scientists understand more about how these viruses recently evolved. These insights may lead to strategies for preventing new coronaviruses from emerging and spreading among humans.
© 2024, Maestri, Perez-Lamarque et al.
Conflict of interest statement
RM, BP, AZ, HM No competing interests declared
Figures















Update of
- doi: 10.1101/2023.03.09.531875
- doi: 10.7554/eLife.91745.1
- doi: 10.7554/eLife.91745.2
References
-
- Adams MJ, Lefkowitz EJ, King AMQ, Harrach B, Harrison RL, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Mushegian AR, Nibert ML, Sabanadzovic S, Sanfaçon H, Siddell SG, Simmonds P, Varsani A, Zerbini FM, Orton RJ, Smith DB, Gorbalenya AE, Davison AJ. 50 years of the International Committee on Taxonomy of Viruses: progress and prospects. Archives of Virology. 2017;162:1441–1446. doi: 10.1007/s00705-016-3215-y. - DOI - PubMed
-
- Alekseev KP, Vlasova AN, Jung K, Hasoksuz M, Zhang X, Halpin R, Wang S, Ghedin E, Spiro D, Saif LJ. Bovine-like coronaviruses isolated from four species of captive wild ruminants are homologous to bovine coronaviruses, based on complete genomic sequences. Journal of Virology. 2008;82:12422–12431. doi: 10.1128/JVI.01586-08. - DOI - PMC - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
