

Inhibition of TOPORS ubiquitin ligase augments the efficacy of DNA hypomethylating agents through DNMT1 stabilization
- PMID: 39198387
- PMCID: PMC11358161
- DOI: 10.1038/s41467-024-50498-4
Inhibition of TOPORS ubiquitin ligase augments the efficacy of DNA hypomethylating agents through DNMT1 stabilization
Abstract
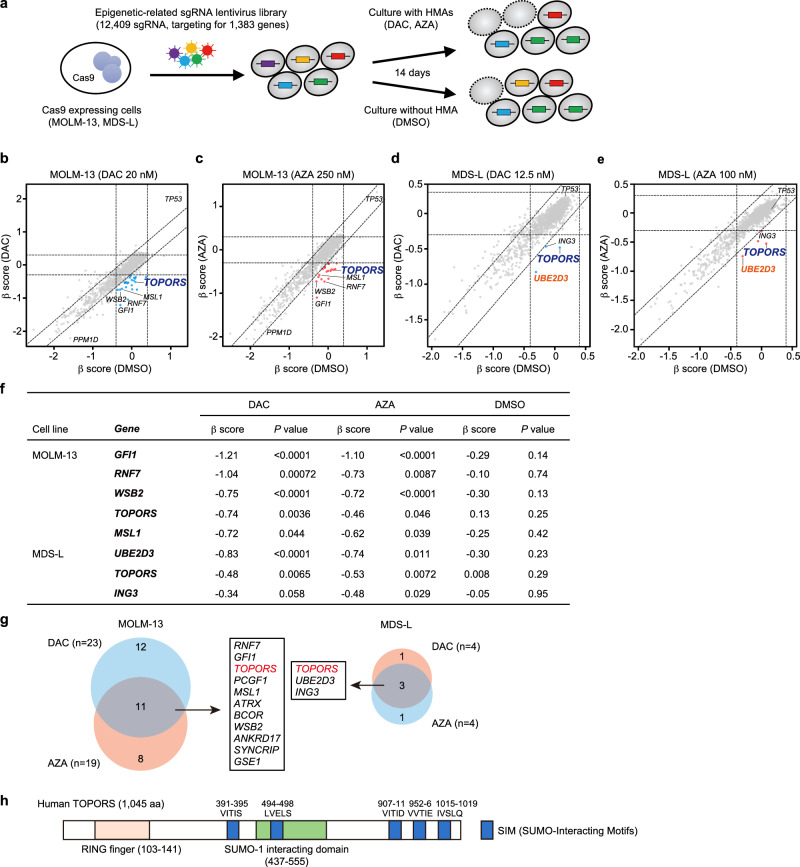
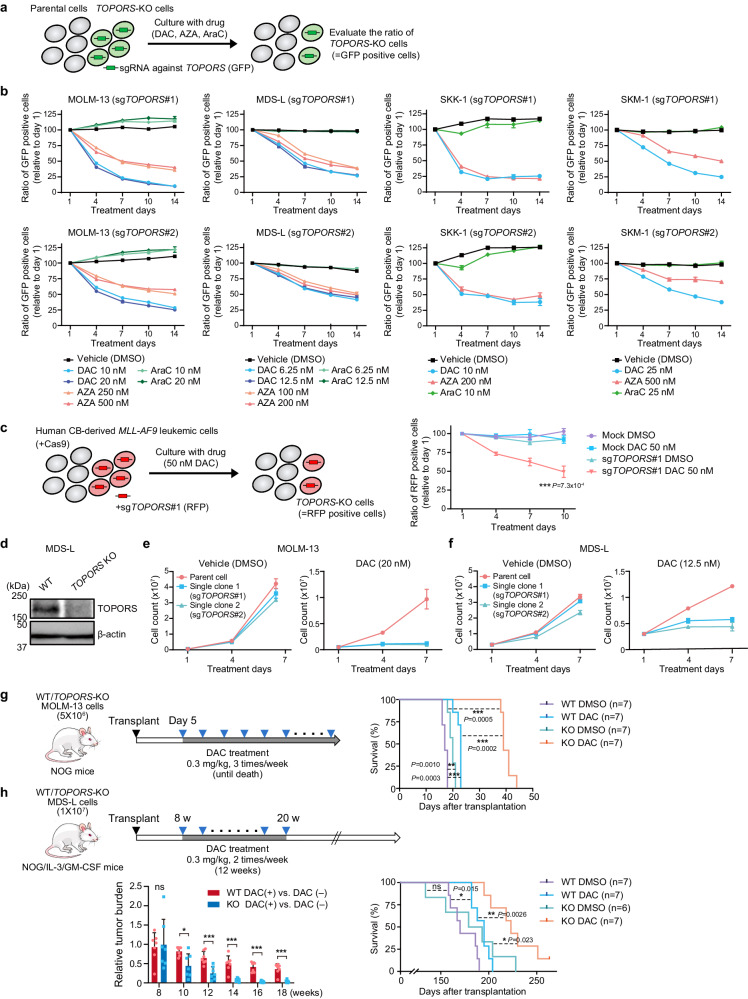
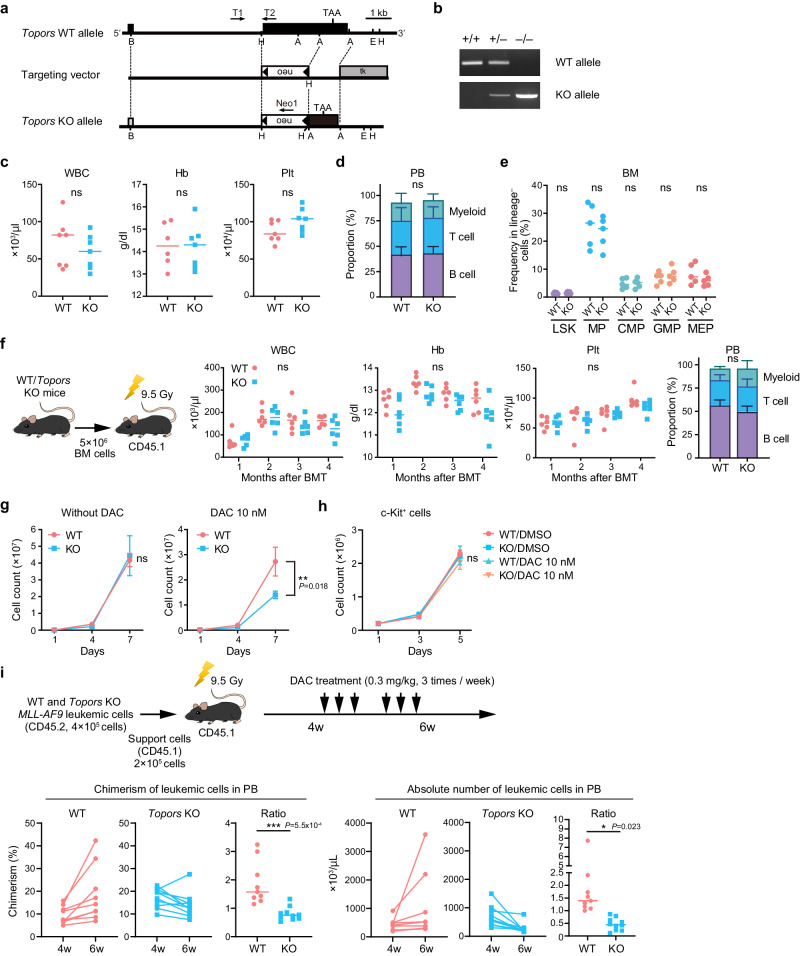
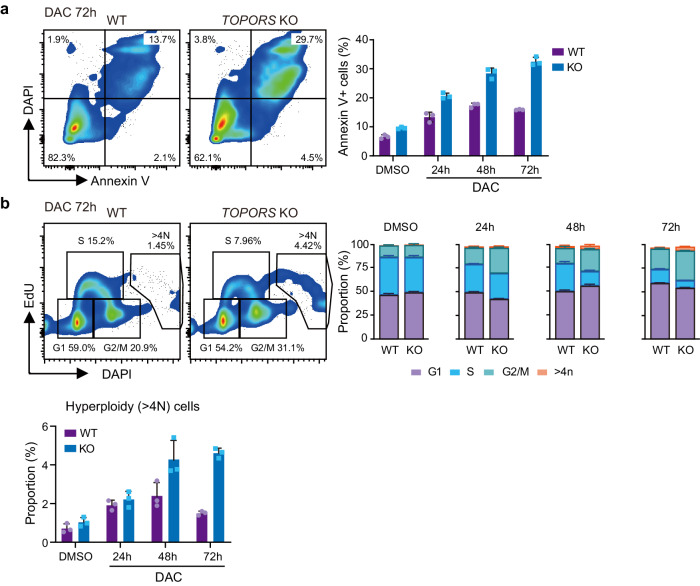
DNA hypomethylating agents (HMAs) are used for the treatment of myeloid malignancies, although their therapeutic effects have been unsatisfactory. Here we show that CRISPR-Cas9 screening reveals that knockout of topoisomerase 1-binding arginine/serine-rich protein (TOPORS), which encodes a ubiquitin/SUMO E3 ligase, augments the efficacy of HMAs on myeloid leukemic cells with little effect on normal hematopoiesis, suggesting that TOPORS is involved in resistance to HMAs. HMAs are incorporated into the DNA and trap DNA methyltransferase-1 (DNMT1) to form DNA-DNMT1 crosslinks, which undergo SUMOylation, followed by proteasomal degradation. Persistent crosslinking is cytotoxic. The TOPORS RING finger domain, which mediates ubiquitination, is responsible for HMA resistance. In TOPORS knockout cells, DNMT1 is stabilized by HMA treatment due to inefficient ubiquitination, resulting in the accumulation of unresolved SUMOylated DNMT1. This indicates that TOPORS ubiquitinates SUMOylated DNMT1, thereby promoting the resolution of DNA-DNMT1 crosslinks. Consistently, the ubiquitination inhibitor, TAK-243, and the SUMOylation inhibitor, TAK-981, show synergistic effects with HMAs through DNMT1 stabilization. Our study provides a novel HMA-based therapeutic strategy that interferes with the resolution of DNA-DNMT1 crosslinks.
© 2024. The Author(s).
Conflict of interest statement
K.H. is a co-founder of Dania Therapeutics, a consultant for Inthera Bioscience AG, and a scientific advisor for MetaboMed Inc. and Hannibal Innovation. All other authors declare no competing interests.
Figures
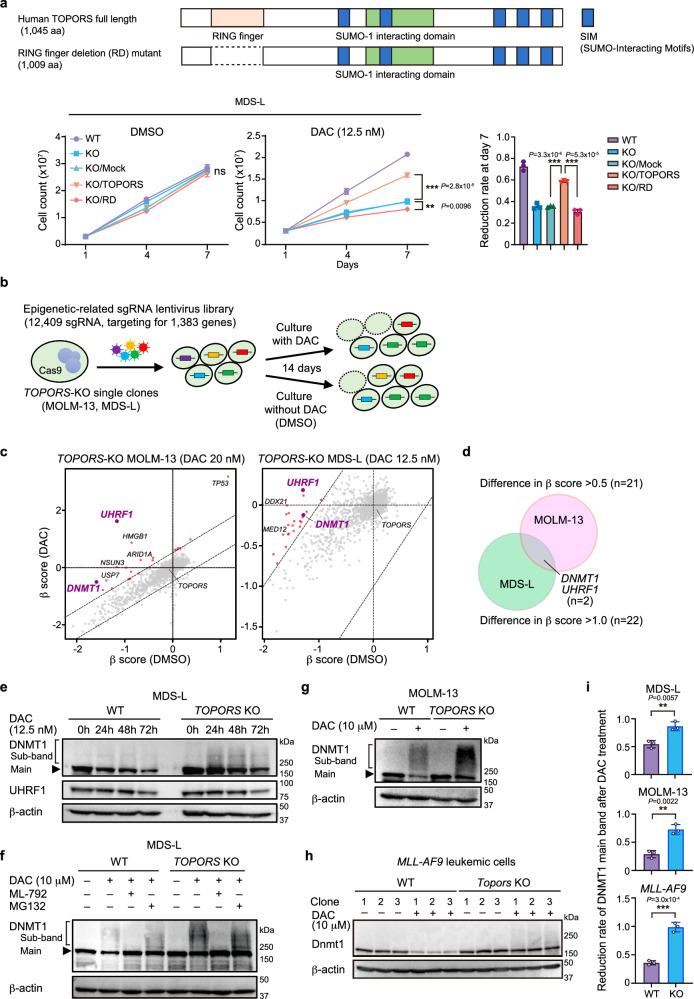
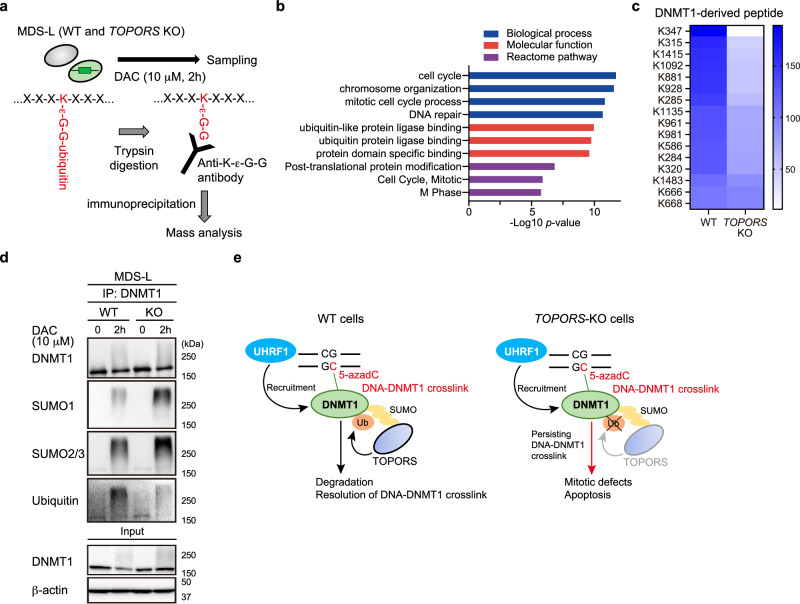
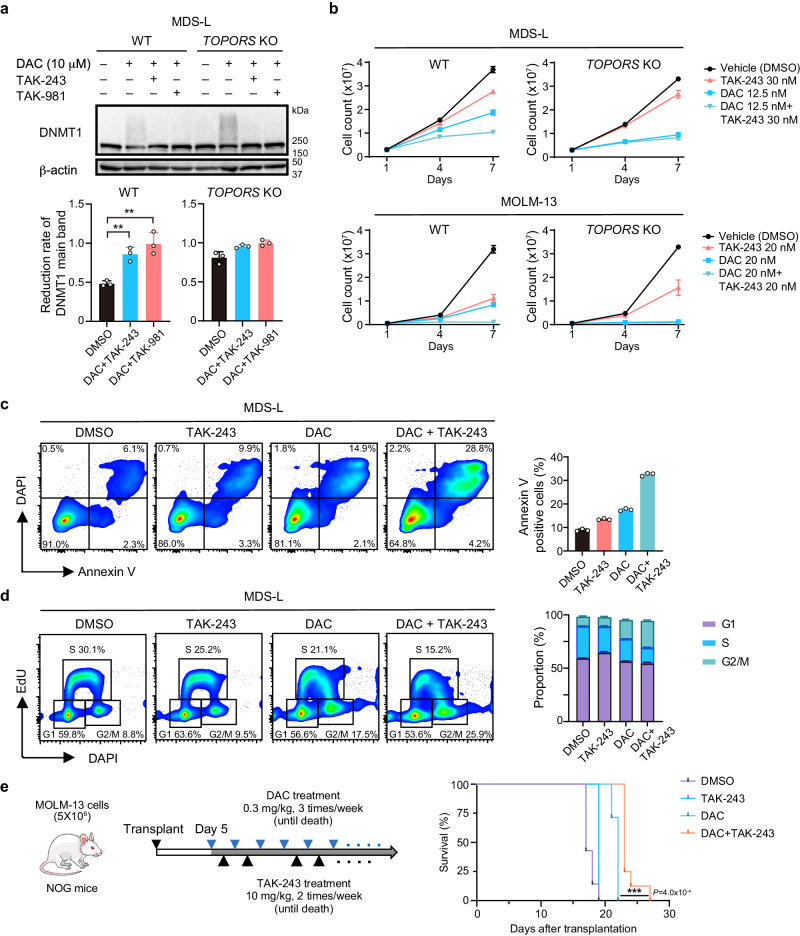
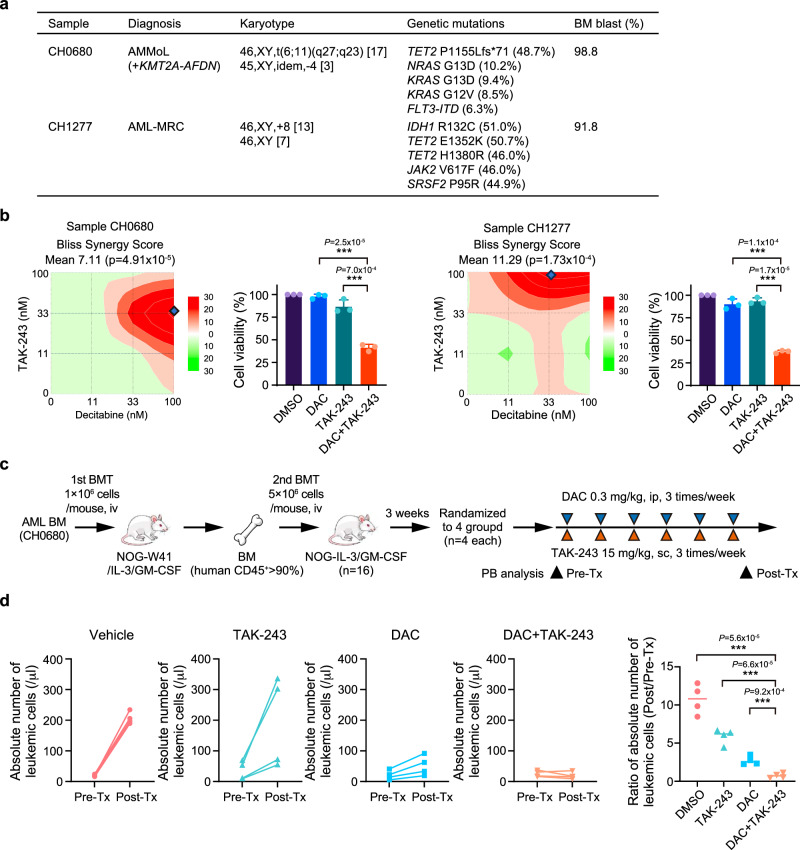
References
-
- Weiss, A. J., Stambaugh, J. E., Mastrangelo, M. J., Laucius, J. F. & Bellet, R. E. Phase I study of 5-azacytidine (NSC-102816). Cancer Chemother. Rep.56, 413–419 (1972). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
