

Systemic inflammation following traumatic injury and its impact on neuroinflammatory gene expression in the rodent brain
- PMID: 39198925
- PMCID: PMC11360339
- DOI: 10.1186/s12974-024-03205-5
Systemic inflammation following traumatic injury and its impact on neuroinflammatory gene expression in the rodent brain
Abstract
Background: Trauma can result in systemic inflammation that leads to organ dysfunction, but the impact on the brain, particularly following extracranial insults, has been largely overlooked.
Methods: Building upon our prior findings, we aimed to understand the impact of systemic inflammation on neuroinflammatory gene transcripts in eight brain regions in rats exposed to (1) blast overpressure exposure [BOP], (2) cutaneous thermal injury [BU], (3) complex extremity injury, 3 hours (h) of tourniquet-induced ischemia, and hind limb amputation [CEI+tI+HLA], (4) BOP+BU or (5) BOP+CEI and delayed HLA [BOP+CEI+dHLA] at 6, 24, and 168 h post-injury (hpi).
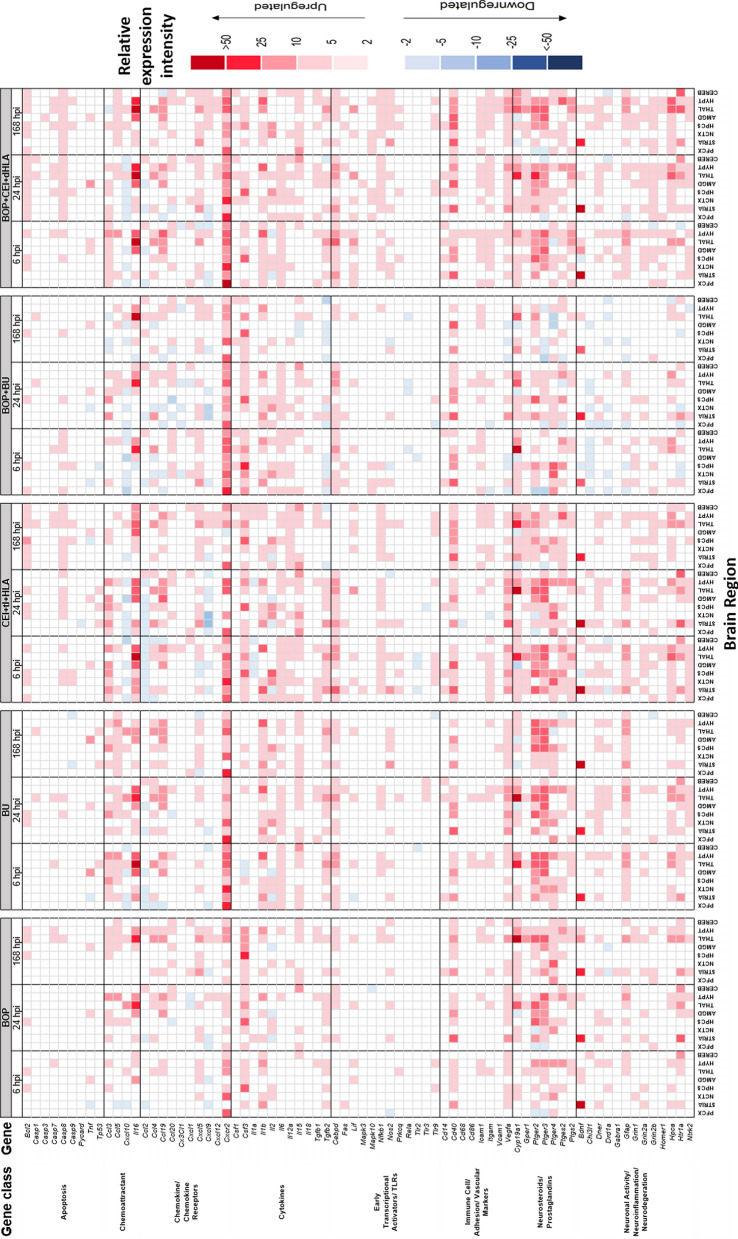
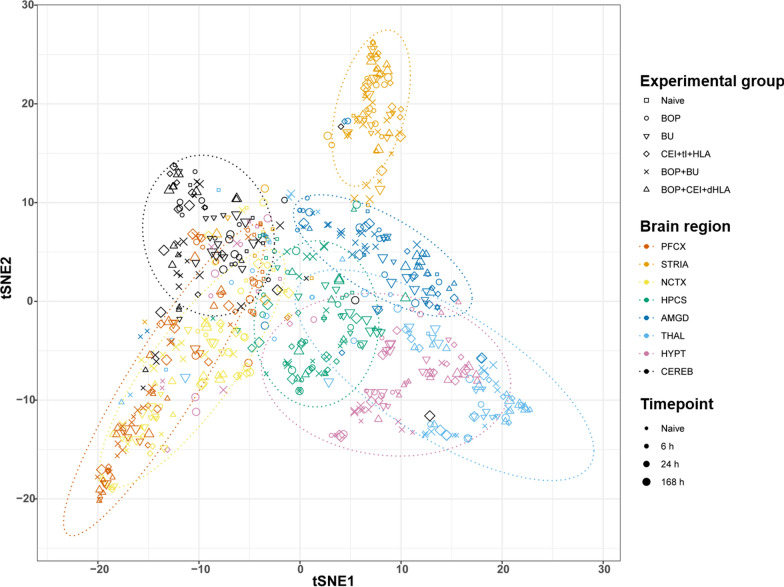
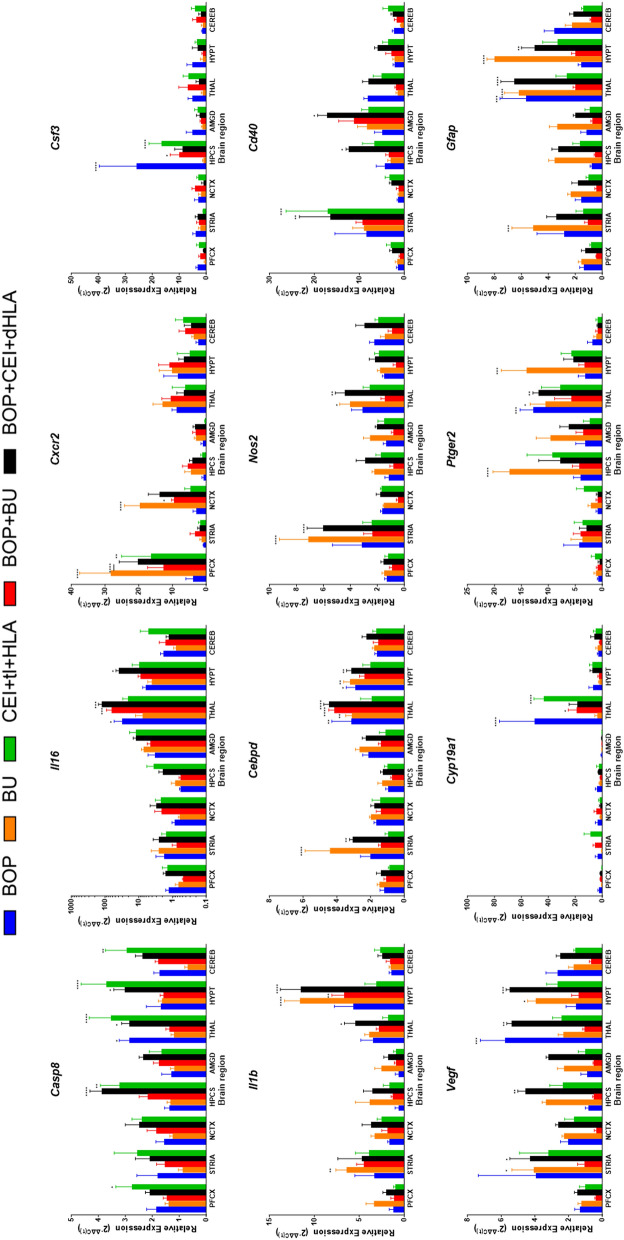
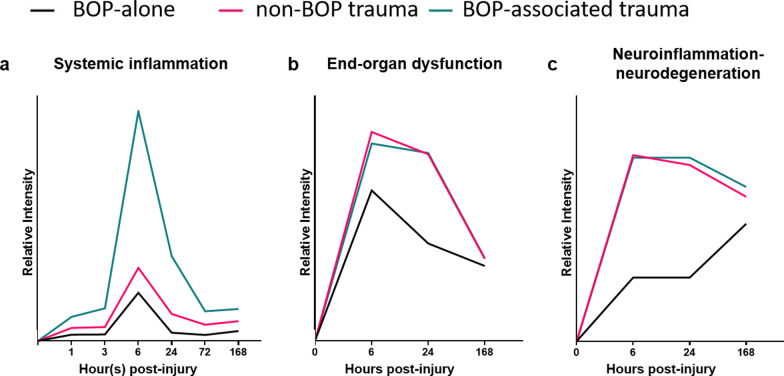
Results: Globally, the number and magnitude of differentially expressed genes (DEGs) correlated with injury severity, systemic inflammation markers, and end-organ damage, driven by several chemokines/cytokines (Csf3, Cxcr2, Il16, and Tgfb2), neurosteroids/prostaglandins (Cyp19a1, Ptger2, and Ptger3), and markers of neurodegeneration (Gfap, Grin2b, and Homer1). Regional neuroinflammatory activity was least impacted following BOP. Non-blast trauma (in the BU and CEI+tI+HLA groups) contributed to an earlier, robust and diverse neuroinflammatory response across brain regions (up to 2-50-fold greater than that in the BOP group), while combined trauma (in the BOP+CEI+dHLA group) significantly advanced neuroinflammation in all regions except for the cerebellum. In contrast, BOP+BU resulted in differential activity of several critical neuroinflammatory-neurodegenerative markers compared to BU. t-SNE plots of DEGs demonstrated that the onset, extent, and duration of the inflammatory response are brain region dependent. Regardless of injury type, the thalamus and hypothalamus, which are critical for maintaining homeostasis, had the most DEGs. Our results indicate that neuroinflammation in all groups progressively increased or remained at peak levels over the study duration, while markers of end-organ dysfunction decreased or otherwise resolved.
Conclusions: Collectively, these findings emphasize the brain's sensitivity to mediators of systemic inflammation and provide an example of immune-brain crosstalk. Follow-on molecular and behavioral investigations are warranted to understand the short- to long-term pathophysiological consequences on the brain, particularly the mechanism of blood-brain barrier breakdown, immune cell penetration-activation, and microglial activation.
Keywords: Blast; Differential gene expression; Neuroinflammation; Polysystem injury; Secondary brain injury.
© 2024. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




References
-
- Magnus D, Khan MA, Proud WG. Epidemiology of civilian blast injuries inflicted by terrorist bombings from 1970–2016. Def Technol. 2018;14:469–76. 10.1016/j.dt.2018.07.014 - DOI
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
