

Risk Assessment and Personalized Treatment Options in Inherited Dilated Cardiomyopathies: A Narrative Review
- PMID: 39200108
- PMCID: PMC11351202
- DOI: 10.3390/biomedicines12081643
Risk Assessment and Personalized Treatment Options in Inherited Dilated Cardiomyopathies: A Narrative Review
Abstract
Considering the worldwide impact of heart failure, it is crucial to develop approaches that can help us comprehend its root cause and make accurate predictions about its outcome. This is essential for lowering the suffering and death rates connected with this widespread illness. Cardiomyopathies frequently result from genetic factors, and the study of heart failure genetics is advancing quickly. Dilated cardiomyopathy (DCM) is the most prevalent kind of cardiomyopathy, encompassing both genetic and nongenetic abnormalities. It is distinguished by the enlargement of the left ventricle or both ventricles, accompanied by reduced contractility. The discovery of the molecular origins and subsequent awareness of the molecular mechanism is broadening our knowledge of DCM development. Additionally, it emphasizes the complicated nature of DCM and the necessity to formulate several different strategies to address the diverse underlying factors contributing to this disease. Genetic variants that can be transmitted from one generation to another can be a significant contributor to causing family or sporadic hereditary DCM. Genetic variants also play a significant role in determining susceptibility for acquired triggers for DCM. The genetic causes of DCM can have a large range of phenotypic expressions. It is crucial to select patients who are most probable to gain advantages from genetic testing. The purpose of this research is to emphasize the significance of identifying genetic DCM, the relationships between genotype and phenotype, risk assessment, and personalized therapy for both those affected and their relatives. This approach is expected to gain importance once treatment is guided by genotype-specific advice and disease-modifying medications.
Keywords: genotype-specific therapy; inherited dilated cardiomyopathy; risk assessment.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
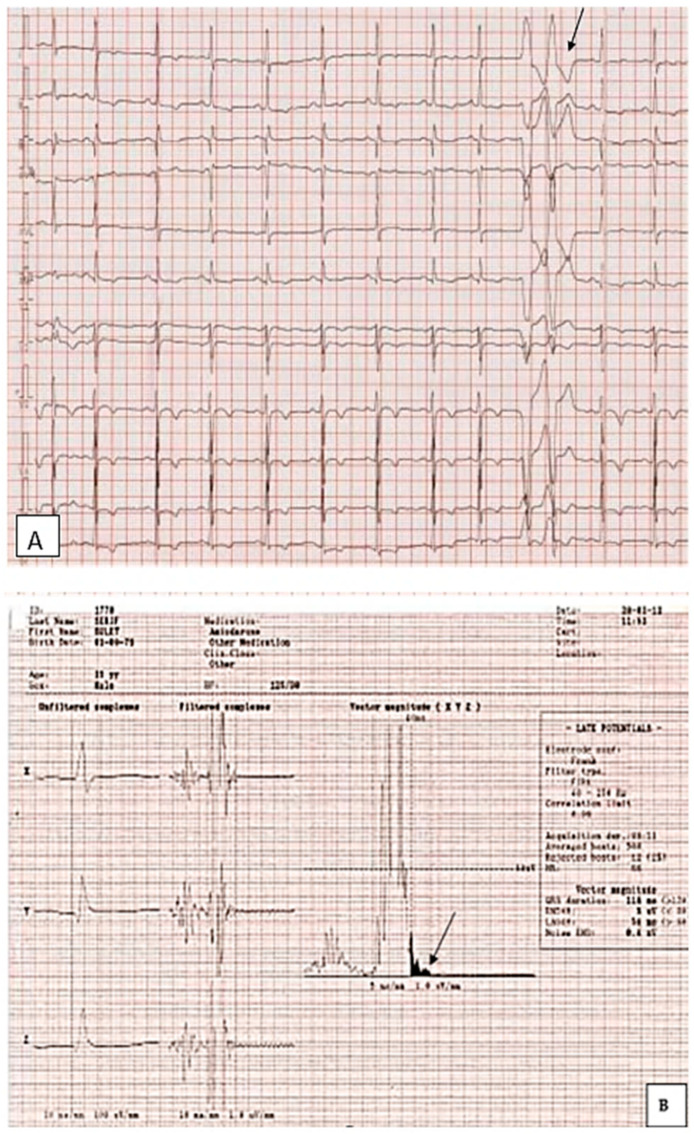
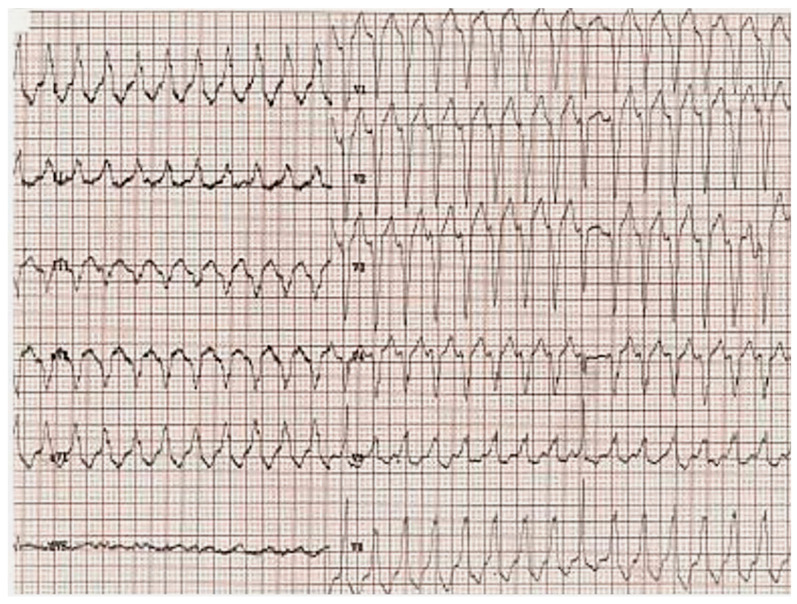
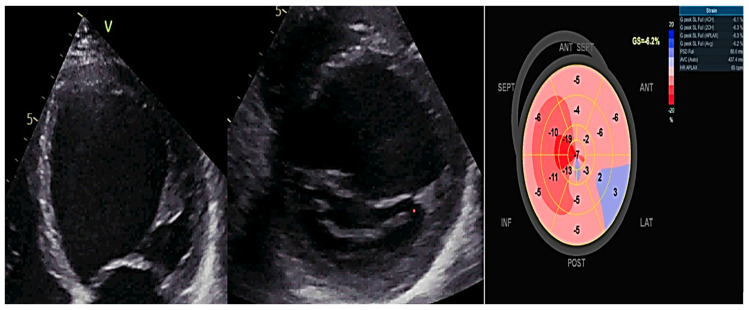
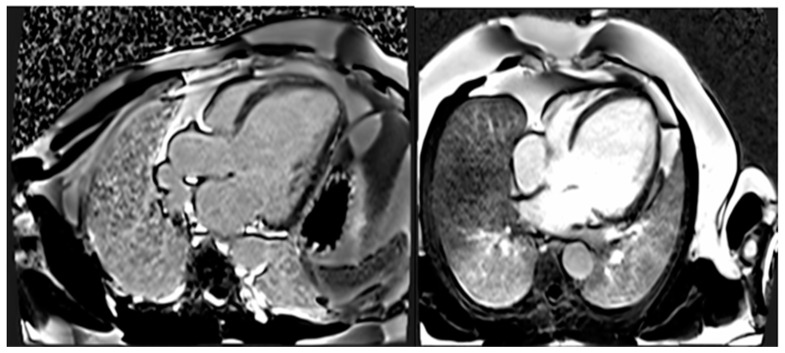
References
-
- Stehlik J., Edwards L.B., Kucheryavaya A.Y., Benden C., Christie J.D., Dobbels F., Kirk R., Rahmel A.O., Hetrz M.I. The Registry of the International Society for Heart and Lung Transplantation: Twenty-eighth Adult Heart Transplant Report—2011. J. Heart Lung Transplant. 2011;30:1078–1094. doi: 10.1016/j.healun.2011.08.003. - DOI - PubMed
-
- Heidenreich P.A., Albert N.M., Allen L.A., Bluemke D.A., Butler J., Fonarow G.C., Ikonomidis J.S., Khavjou O., Konstam M.A., Maddox T.M., et al. Forecasting the impact of heart failure in the United States a policy statement from the American Heart Association. Circ. Heart Fail. 2013;6:606–619. doi: 10.1161/HHF.0b013e318291329a. - DOI - PMC - PubMed
-
- Arbelo E., Protonotarios A., Gimeno J.R., Arbustini E., Barriales-Villa R., Basso C., Bezzina C.R., Biagini E., Blom N.A., de Boer R.A., et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC) Eur. Heart J. 2023;44:3503–3626. doi: 10.1093/eurheartj/ehad194. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
